陈 计,高 鹏,陆兆新,吕凤霞,张 充,赵海珍,别小妹 *
(南京农业大学食品科技学院,江苏 南京 210095)
摘 要:为获得β-1,3-1,4-葡聚糖酶高产菌株,以高地芽孢杆菌YC-9为出发菌株,通过亚硝基胍(nitrosoguanidine,NTG)和低能N +束诱变育种,筛选和选育得到两株突变株N-2-2 和10-30s-3,发酵培养60 h,酶 活力分别达到28.6 U/mL和36.1 U/mL ,分别是出发菌株的2.36 倍和2.98 倍,与YC-9菌株相比,突变菌株菌体生长下降但发酵产酶量增加。进一步将β-1,3-1,4-葡聚糖酶克隆并在大肠杆菌中成功表达,经过30 ℃诱导6 h后,胞内酶活力达79.2 U/mL,为出发菌株的6.5 倍。
关键词:高地芽孢杆菌;β-1,3-1,4-葡聚糖酶;亚硝基胍诱变;低能N +束注入;异源表达
β-1,3-1,4-葡聚糖酶(β-1,3-1,4-D-葡聚糖-4-葡萄糖水解酶,E.C.3.2.1.73)分解β-1,3-1,4-葡聚糖中与β-D-1,3糖苷键相邻的β-D-1,4-糖苷键,降解产物主要是纤维三糖和纤维四糖,因其主要分解大麦中的β-1,3-1,4-葡聚糖和细菌地衣多糖,所以又称地衣多糖酶 [1]。β-1,3-1,4-葡聚糖酶是一种重要的工业用酶,在啤酒酿造中,主要用于改善啤酒的混浊度,提高产品质量 [2],在饲料工业中,主要用作饲料添加剂,可有利于动物对营养物质的消化和吸收,提高生长性能和粮食的转化率 [3]。迄今为止,β-1,3-1,4-葡聚糖酶已在芽孢杆菌、曲霉、毛霉、青霉和瘤胃微生物中得到广泛的发现 [4-7],然而大部分原始菌株的β-1,3-1,4-葡聚糖酶的酶活力或者热稳定性较低,这就限制了其在工业上的应用。现阶段,提高酶表达量或者酶活的方法主要有:遗传育种、异源表达以及酶分子的定向和非定向改造。其中传统诱变育种因其成本低廉、方法简便、效果显著等优点,至今仍在生产实践中广泛应用 [8-10]。而通过分子生物学手段,将外源酶基因导入宿主菌,构建异源表达系统,已成为当今科研界提高酶表达量的基本策略。
本课题组Mao Shurui等 [11]在前期筛选和鉴定了产β-1,3-1,4-葡聚糖酶的高地芽孢杆菌YC-9,但原始菌株的酶表达量较低。本研究以高地芽孢杆菌YC-9为出发菌株,对其进行了诱变育种,并将β-1,3-1,4-葡聚糖酶基因在大肠杆菌中进行了异源表达。
1.1 菌种、培养基与试剂
高地芽孢杆菌YC-9(Bacillus altitudinis YC-9,保藏号CGMCC4314)、 Escherichia coli DH5α克隆宿主与Escherichia coli BL21(DE3) pLysS表达宿主为本实验室保藏。
营养琼脂(NA)培养基:牛肉膏3 g、蛋白胨10 g、NaCl 5 g、琼脂15 g,蒸馏水1 000 mL,pH 7.2;LB培养基(液体):酵母膏5 g、胰蛋白胨10 g、NaCl 10 g,蒸馏水1 000 mL,pH 7.0;初筛培养基:酵母膏5 g、胰蛋白胨10 g、NaCl 10 g、地衣多糖1 g、刚果红0.4 g,蒸馏水1 000 mL,pH 7.0。
克隆载体p M D 1 9-T载体、限制性内切酶BamHⅠ、XhoⅠ、Taq DNA聚合酶、T4 DNA连接酶、X-Gal、异丙基硫代-β-D-半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG) 宝生物工程(大连)有限公司;表达载体pET-28a(+) 南京大学生命科学学院房云彬所赠;地衣多糖(lichenan) 爱尔兰Megazyme公司;X-Gal/IPTG Premix、dNTP、DNA标准分子质量Marker 南京金斯瑞公司;DNA凝胶回收试剂盒、柱式质粒提取试剂盒、琼脂糖、十二烷基硫酸钠(sodium dodecyl sulfate,SDS) 生工生物工程(上海)股份有限公司;酵母提取物、胰蛋白胨等 英国Oxoid公司;其他试剂均为市售分析纯。
1.2 仪器与设备
UV-2450紫外-可见分光光度计 日本岛津公司;Centrifuge 5804R冷冻离心机 德国Eppendorf公司;Orion 3 STAR pH计 美国Orion公司;GXZ-9240 MBE恒温干燥箱、PYX-DHS-50X65隔水式电热恒温培养箱上海跃进医疗器械厂;JS-380C全自动数码凝胶成像分析仪 上海培清科技有限公司;PTC-100 TMPCR热循环仪、PowerPac TMHC电泳仪 美国Bio-Rad公司。
1.3 方法
1.3.1 β-1,3-1,4-葡聚糖酶活性测定
采用3,5-二硝基水杨酸(3,5-dinitrosalicylic acid,DNS)法 [12]。酶活力单位定义为:在60 ℃、pH 7.0条件下每分钟从底物中释放1 μmol还原糖(以葡萄糖计)所需的酶量为一个酶活力单位(U)。计算公式如下:
式中:X为样品的酶活力/(U/mL);n为稀释倍数;ρ为由回归方程计算出的葡萄糖的含量/(mg/mL);10为反应时间,取10 min;M表示葡萄糖的分子质量/(g/mol)。
1.3.2 高地芽孢杆菌的亚硝基胍(nitrosoguanidine,NTG)诱变
称取20 mg固体NTG溶于10 mL丙酮,使NTG溶液初始质量浓度为2 mg/mL。分别取NTG母液0.20、0.15、0.12、0.10、0.08、0.06、0.04、0.02、0 mL加入9 个4 mL离心管中,再吸取1 mL菌液加到各个离心管里,反应体系为2 mL,体积不够2 mL的用pH 6的缓冲液补足。37 ℃反应30 min后取100 μL稀释菌液涂布平板,37 ℃培养、挑单菌落、初筛、复筛。
1.3.3 高地芽孢杆菌的低能N +束注入诱变
本实验所用的离子束辐照设备由南京工业大学提供,采用低能N +注入,注入能量为10 keV和20 keV。将需离子注入的样品培养皿叠放在对照培养皿上方,一起放入离子注入机靶室,打开上方培养皿盖,靶室抽真空。在两个能量条件下,放置时间分别是0、30、60、90 s。随后吸取1 mL无菌水洗脱诱变后的菌膜成为1 mL菌液,取100 μL稀释菌液涂布平板,37 ℃培养、挑单菌落、初筛、复筛。
1.3.4 初筛和复筛
取100 μL稀释菌液涂布平板,37 ℃培养24 h,菌落计数。用灭菌的牙签挑取单菌落点种于初筛平板上,37 ℃培养24 h。用游标卡尺量取菌落周围的水解圈,挑取水解圈大的菌株作为复筛菌株,并分别按公式(2)、(3)计算致死率、突变率 [13]。
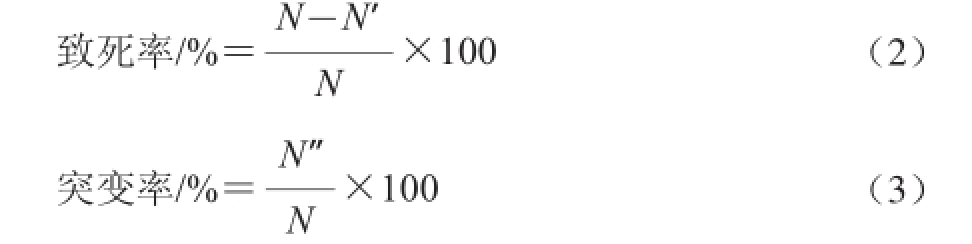
式中:N为对照组单菌落数/(CFU/mL);N′为诱变处理样品单菌落数/(CFU/mL);N”为突变菌落数/(CFU/mL)。
将经初筛得到的菌株于50 mL LB液体培养基中37 ℃条件下180 r/min摇床培养6 h左右,至OD 600 nm值为0.6~0.8,制备种子液;再按2%接种量将种子液接种至发酵培养基,于37 ℃条件下180 r/min摇床培养60 h。发酵培养液于5 000×g离心15 min,取上清液测酶活力。
1.3.5 遗传稳定性实验
将诱变筛选出的高产菌株进行传代培养,每隔24 h传代1 次,共传6 代,然后将每一代菌株分别进行摇瓶发酵,根据酶活大小检测菌株的传代稳定性。
1.3.6 诱变菌株与原始菌株的生长产酶情况比较
将经NTG诱变和低能N +束诱变得到的菌株N-2-2和10-30s-3,以及原始菌种高地芽孢杆菌YC-9分别接种于100 mL LB液体培养基,于37 ℃条件下180 r/min摇床培养6 h左右至OD 600 nm值为0.6~0.8,制备种子液;再按2%接种量将种子液接种至发酵培养基,于37 ℃条件下180 r/min摇床培养60 h,每隔6 h取样3 mL,于紫外-可见分光光度计600 nm波长处测菌体浓度,同时将所取样品于5 000×g离心15 min,取上清液测酶活力,比较诱变后得到的菌株与原始菌株的生长产酶情况差异。
1.3.7 β-1,3-1,4-葡聚糖酶基因的扩增
根据GenBank登录的高地芽孢杆菌YC-9 β-1,3-1,4-葡聚糖酶的基因序列,用引物设计软件Primer premier 5及DNAMAN等生物软件,设计β-1,3-1,4-葡聚糖酶扩增引物:上游引物bgl-F:5′-CGGATCCATGTCTTACCGTGT GAAACGAATG-3′,下游引物bgl-R:5′-CCGCTCGAG TTATCTTTTTGTGTAACGTACC-3′,下划线分别为酶切位点BamHⅠ和XhoⅠ,酶切位点前为保护碱基。以高地芽孢杆菌YC-9基因组为模板,以bgl-F、bgl-R为引物,扩增β-1,3-1,4-葡聚糖酶基因,聚合酶链式反应(polymerase chain reaction,PCR)反应程序:94 ℃预变性2 min;94 ℃变性30 s;55 ℃退火30 s;72 ℃延伸1 min;30 个循环;72 ℃延伸10 min;PCR产物经过1.0%琼脂糖回收预期大小的片段,与pMD19-T载体连接,转化入大肠杆菌DH5α感受态细胞,涂布在含氨苄青霉素LB平板上,挑选阳性克隆,提质粒进行测序验证,得重组克隆载体pMD19-T-bgl。
1.3.8 重组表达载体的构建
将获得的pMD19-T-bgl用BamHⅠ和XhoⅠ进行双酶切,回收目的片段,并与同样经过BamHⅠ和XhoⅠ双切的载体pET-28a(+)进行16 ℃过夜连接,连接产物转化大肠杆菌BL21感受态细胞,涂布于含50 μg/mL卡那霉素(Kan)的LB平板,37 ℃过夜培养后。挑选阳性克隆子提取质粒进行测序,并用BamHⅠ和XhoⅠ进行双酶切验证。
1.3.9 SDS-聚丙烯酰胺凝胶电泳(polyacrylamide gelelectrophoresis,PAGE)电泳检测β-1,3-1,4-葡聚糖酶在E. coli BL21中的表达
将β-1,3-1,4-葡聚糖酶基因重组菌株E. coli BL21(pET-28a-bgl)和参照菌株E. coli BL21(pET-28a(+)),分别接种一环到加有50 μg/mL Kan的液体LB培养基中,置于37 ℃条件下180 r/min摇床培养过夜;然后按2%的接种量转接到100 mL含有相同量Kan的新鲜液体LB培养基中,37 ℃、180 r/min摇床培养至菌体质量浓度0.6~0.8 μg/mL,然后加入IPTG至终质量浓度为100 μg/mL,37 ℃、180 r/min诱导培养6 h,离心弃上清,用细胞破碎液悬浮细胞,置于细胞破碎仪下破碎,破碎完,离心取上清,进行SDS-PAGE电泳。采用质量分数12%分离胶,质量分数5%浓缩胶的SDS-PAGE,样品加入5×上样缓冲液,沸水浴5 min后,上样15 μL进行电泳。BL21菌株除了不加抗生素外,其他操作与重组菌株和参照菌株一致。
1.3.10 不同温度下工程菌诱导产酶情况
将β-1,3-1,4-葡聚糖酶基因重组菌株E. coli BL21(pET-28a-bgl)接种一环到加有50 μg/mL Kan的液体LB培养基中,置于37 ℃、180 r/min摇床培养过夜;然后按2%的接种量转接到100 mL含有相同量Kan的新鲜液体LB培养基中,37 ℃、180 r/min摇床培养至菌体OD 600 nm值为0.6~0.8,然后加入IPTG至终质量浓度为100 μg/mL,分别于37、30、16 ℃条件下诱导培养6 h。离心弃上清,用细胞破碎液悬浮细胞,置于细胞破碎仪下破碎,破碎完,离心取上清液,测酶活力。
2.1 NTG诱变后的致死率、正负突变率及复筛结果
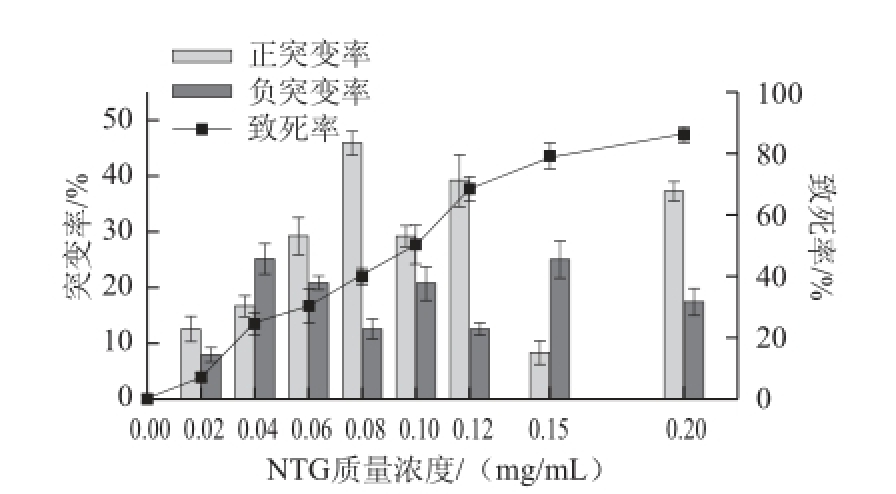
图1 NTG诱变后的致死率和正负突变率
Fig.1 Mortality, positive and negative mutation rates after NTG mutagenesis
由图1可知,随着NTG质量浓度的升高,致死率不断提高,正突变率整体呈现先上升后下降的趋势,并在NTG质量浓度为0.08 mg/mL时最大,因此确定该质量浓度为后续NTG诱变时的最佳处理条件。经过两轮诱变筛选,获得一株产β-1,3-1,4-葡聚糖酶酶活性较原始菌株有较大提高的菌株,并命名为N-2-2,酶活力为(28.6±1.2)U/mL,较原始菌株酶活力((12.1±1.3)U/mL)提高136%。
2.2 低能N +束注入诱变后的致死率及复筛结果

图2 N 2 N
+束注入诱变处理结果
Fig.2 Mortality after N
+beam implantation
由图2可知,在注入能量为20 keV条件下,注入30 s时,致死率已高达80%以上,说明该能量条件下,注入较短时间,已产生较大的致死效应,因此不适合本实验菌株的诱变;当注入能量为10 keV时,在注入时间分别为30、60、90 s时,致死率分别为10.1%、21.5%、74.7%,随着注入时间的延长,致死率不断增加,可以看出此注入能量适合本实验菌株的诱变。本次N +注入诱变的出发菌株为NTG诱变后获得的菌株N-2-2,经过一轮N +注入诱变,在注入能量为10 keV,注入时间为30 s条件下,筛选出一株产β-1,3-1,4-葡聚糖酶酶活力较出发菌株N-2-2提高26.2%的菌株,命名为10-30s-3,该菌株酶活力达(36.1±2.5)U/mL,是原始菌株的2.98 倍。
2.3 遗传稳定性实验

图3 突变株遗传稳定性
Fig.3 Genetic stability of the two mutants and the original strain
由图3可知,摇瓶发酵时,诱变菌株N-2-2、10-30s-3的β-1,3-1,4-葡聚糖酶酶活力一直高于原始菌株,在传6 代时,酶活力都维持在相对稳定的范围,能够较稳定遗传,说明诱变提高了β-1,3-1,4-葡聚糖酶的产量。
2.4 诱变菌株与原始菌株的生长产酶情况比较
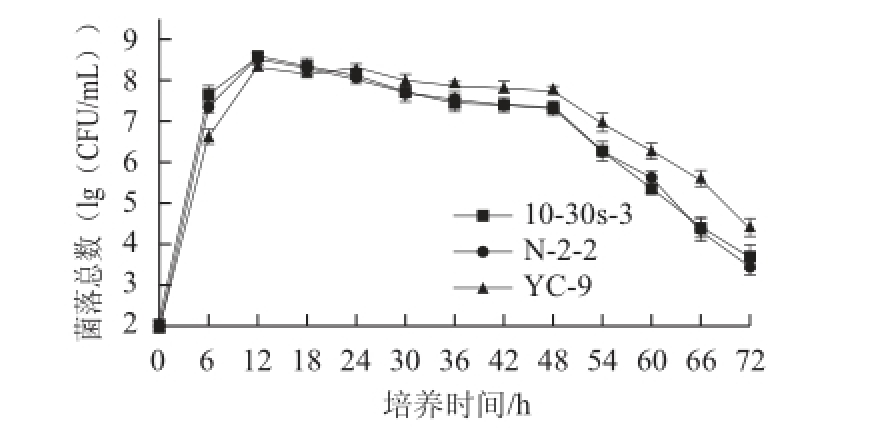
图4 菌株N-2-2、10-30s-3及原始菌株生长情况比较
Fig.4 Comparison of the growth characteristics of the mutant strains and the parent strain
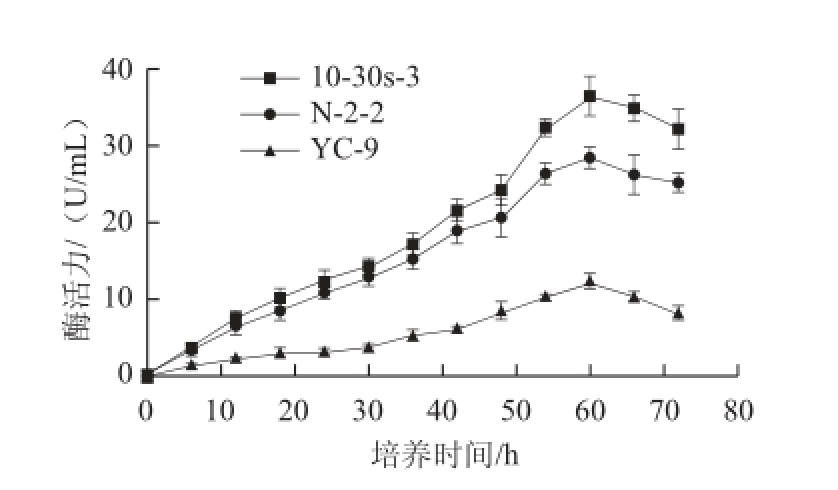
图5 菌株N-2-2、10-30s-3及原始菌株产酶情况比较
Fig.5 Comparison of the enzyme production of the mutant strains and the parent strain
由图4可知,3 株菌株的生长情况大致可分为以下3 个阶段,在0~12 h处于对数生长期,在12~48 h处于相对稳定期,在48~72 h处于明显衰亡期。在对数期,诱变菌株N-2-2和10-30s-3的生长速率明显高于原始菌株YC-9;在稳定期,3 株菌株活菌数均在下降,但诱变菌株N-2-2和10-30s-3的下降趋势较原始菌株YC-9要更明显;在培养24 h后,诱变菌株N-2-2和10-30s-3的活菌数始终低于原始菌株YC-9。由图5可知,诱变菌株和原始菌株的产酶特征较一致,均随着时间的延长,酶活力不断增加,并且大致都在发酵60 h左右,酶活力达到最大值;但每一取样点,诱变菌株的酶活力都高于原始菌株;综上,突变菌株与原始菌株在生长和产酶上有着较大的差异,即突变菌株生长速率高于原始菌株,且下降趋势更快,除此之外,突变菌株在生长不断下降的时候,酶活力上升趋势却快于原始菌株。
2.5 β-1,3-1,4-葡聚糖酶基因的扩增

图6 6 β-1,3-1,4-葡聚糖酶基因的扩增
Fig.6 Amplifi cation of β-1,3-1,4-glucanase gene
1. 阴性对照;2、3. 分别以bgl-F、bgl-R为引物扩增的β-1,3-1,4-葡聚糖酶基因;4. DL2000 Marker。
利用引物bgl-F/bgl-R扩增得到的阳性条带大小在750 bp左右,如图6所示,与目的基因β-1,3-1,4-葡聚糖酶的大小(732 bp,编码243 个氨基酸)基本相符,测序验证,成功扩增出β-葡聚糖酶基因。
2.6 重组表达载体的构建
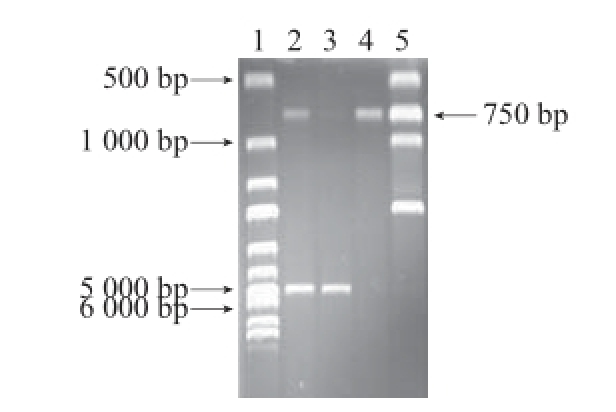
图7 重组质粒pET28a(+)-bgl的双酶切鉴定
Fig.7 Identifi cation of recombinant plasmid pET28a-bgl digested with BamHⅠ and XhoⅠ
1. 1 kb Ladder Marker;2. 重组质粒pET-28a-glu经BamHⅠ和XhoⅠ双酶切;3. pET-28a(+)空载;4. 扩增的β-1,3-1,4-葡聚糖酶基因;5. DL2000 Marker。
将双酶切及测序验证正确的pMD19-T-bgl和载体pET-28a(+)分别经BamHⅠ和XhoⅠ双酶切,回收相应片段,连接产物转化大肠杆菌BL21,挑取转化子扩增并抽提质粒。将抽提的质粒经BamHⅠ和XhoⅠ双酶切,得到了750 bp左右和5 000~6 000 bp左右的条带(图7),证实β-1,3-1,4-葡聚糖酶基因成功插入载体pET-28a(+),将重组质粒命名为pET-28a-bgl。
2.7 SDS-PAGE电泳检测β-1,3-1,4-葡聚糖酶在E. coli BL21中的表达
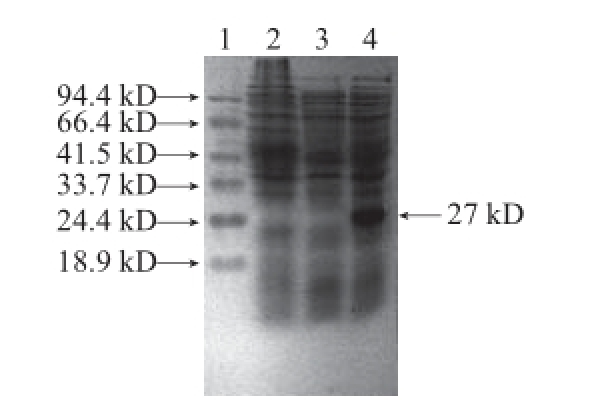
图8 SDS-PAGE电泳检测-1,3-1,4-葡聚糖酶在重组E. coli BL21中的表达
Fig.8 SDS-PAGE analysis of β-1,3-1,4-glucanase expression in recombinant E. coli BL21
1. Marker;2. E.coli BL21(pET-28a);3. E.coli BL21;4. E.coli BL21(pET-28a-bgl)。
由图8可知,工程菌E. coli BL21(pET-28a-bgl)中有一条较深的与27 kD的理论分子质量相近的蛋白条带(箭头所示),而E. coli BL21和E. coli BL21(pET-28a(+))中未见相应的条带,该结果表明β-1,3-1,4-葡聚糖酶重组载体pET-28a-bgl在E. coli BL21中成功实现了表达。Mao Shurui等 [11]用pET-32a(+)也实现了该酶在大肠杆菌中的高效表达,由于pET-32a(+)带有18 kD的融合表达标签(Trx·Tag/His·Tag/S·Tag),使得表达后的酶蛋白以大小45 kD左右的形式出现。而本研究所用表达载体为pET-28a(+),由于不含Trx·Tag/S·Tag等标签,使得所表达的重组蛋白以更接近原始蛋白大小的形式出现。
2.8 不同温度下工程菌E. coli BL21(pET-28a-bgl)诱导产酶情况
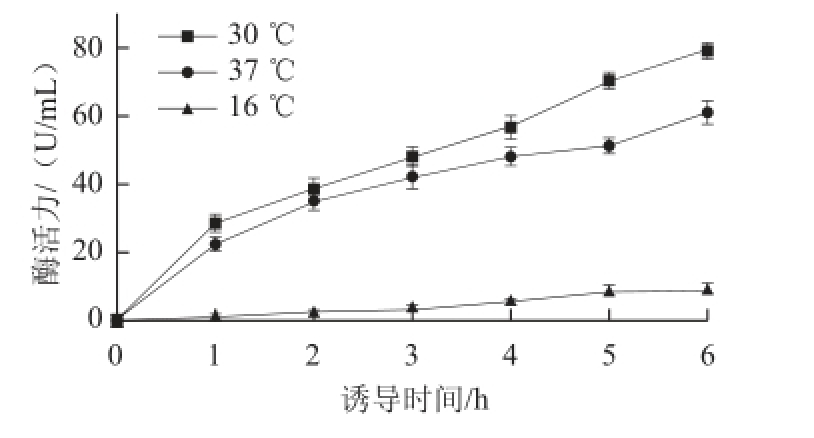
图9 不同温度下工程菌E. coli BL21(pET-28a-bgl)诱导产酶比较
Fig.9 Induced production of the enzyme in E. coli BL21 (pET-28a-bgl) at different temperatures
由图9可知,30 ℃更适合工程菌E. coli BL21(pET-28a-bgl)的诱导产酶,且在诱导6 h后,酶活力达79.2 U/mL,是原始菌株的6.5 倍。
目前,国内外对β-1,3-1,4-葡聚糖酶高产菌株的诱变选育还鲜有报道。本研究利用NTG处理原始菌株,获得了一株β-1,3-1,4-葡聚糖酶高产菌株,酶活力为原始菌株的2.36 倍,结果表明,利用单一诱变剂或诱变方式对出发菌株进行诱变处理,即可获得高产突变株。通常菌株在经过同一诱变剂的多轮处理后会对其产生耐受性,因而若想进一步提高突变株产量,需选择其他诱变剂或诱变方式,在这方面国内外已经取得了良好的结果 [14-15]。因此,本研究在NTG诱变处理的基础上,再用N +束处理获得的突变株,但是效果并不理想,所筛选出的一株酶活力提高最高的突变株,较出发菌株相比,也只提高了26.2%,结果表明,有些情况下,多种诱变剂或诱变方式依次或者复合对菌株进行处理,并不一定能够得到高产菌株,有时甚至会产生较高的负效应 [16]。所有这些都提示诱变育种是一项复杂的科研工作,存在一定的偶然性。进一步实验发现,突变菌株的生长特点和产酶性能与原始菌株有较大的差异,这种诱变后出现的生长代谢差异在其他报道中也有提及,例如,万涛等 [17]利用激光诱变石油脱硫菌 cordonia sp. WQ-01后获得的突变株,延滞期较短,对数期菌体生长更快﹑降解二苯并噻吩(dibenzothiophene,DBT)的速率更高,但对于这些改变的深层次原因尚不清楚,因此有待于进一步的研究。而与传统的诱变育种相比,利用基因工程手段,异源表达酶基因,周期更短,效果更明显 [18-20],更不具有菌株退化甚至回复突变的可能,已成为提高目标物产量的首选。因此,本研究在对原始菌株进行诱变后,又利用基因工程技术,将来自高地芽孢杆菌的β-1,3-1,4-葡聚糖酶基因成功地在大肠杆菌中进行了克隆表达,经诱导,胞内酶活力为原始菌株的6.5 倍,和传统的诱变育种相比,酶活性提高幅度更大。本研究不仅获得了β-1,3-1,4-葡聚糖酶酶活有较大提高的突变株,还获得了高效表达β-1,3-1,4-葡聚糖酶的重组大肠杆菌,这些都为热稳定性β-1,3-1,4-葡聚糖酶的进一步研究和工业化应用打下了坚实的基础。
参考文献:
[1] HRMOVA M, BANIK M, HARVEY A J, et al. Polysaccharide hydrolases in germinated barley and their role in the depolymerization of plant and fungal cell walls[J]. International Journal of Biological Macromolecules, 1997, 21(1): 67-72.
[2] 毕静. β-葡聚糖酶在啤酒生产中的应用研究[J]. 中国酿造, 2011, 30(9): 105-106.
[3] OFFICER D L. Effect of multi-enzyme supplements on the growth performance of piglets during the pre-and post-weaning periods[J]. Animal Feed Science and Technology, 1995, 56(1): 55-65.
[4] SAIT M E, SHELLA I M, HARRY J H. Isolation and over expression of a gene encoding an extracellular β-glucanase from Streptococcusbovis JBI[J]. Applied And Environmental Microboilogy, 1997, 63(10): 3752-3756.
[5] FLINT H J, MEPHERSON E C, BISSET J. Molecular cloning of genes from ruminococcus fl avefaciens encoding xylanase and β-1,3-1,4-glucanase activities[J]. Applied and Environmental Microbiology, 1989, 55(5): 1230-1233.
[6] YANG Shaoqing, QIAO Juanyan, JIANG Zhengqiang, et al. Biochemical characterization of a novel thermostable β-glucanase (lichenase) from Paecilomyces thermophila[J]. Journal of Agricultural and Food Chemistry, 2008, 56(13): 5345-5351.
[7] JUNG Y J, YOO J S, LEE Y S, et al. Purifi cation and characterization of thermostable β-1,3-1,4-glucanase from Bacillus sp. A8-8[J]. Biotechnology and Bioprocess Engineering, 2007, 12(3): 265-270.
[8] 王璋, 王灼维, 莫湘筠, 等. 微生物转谷氨酰胺酶的生产菌种诱变和发酵生产分析[J]. 生物加工过程, 2003, 1(1): 52-59.
[9] 赵彩艳, 李庚飞, 尤跃钧, 等. 氮离子注入选育β-葡聚糖酶高产菌株的研究[J]. 饲料研究, 2008, 4(3): 44-47.
[10] WANG Haiyan, LIU Daimei, LIU Yan, et al. Screening and mutagenesis of a novel Bacillus pumilus strain producing alkaline protease for dehairing[J]. Letters in Applied Microbiology, 2007, 44(1): 1-6.
[11] MAO Shurui, LU Zhaoxin, ZHANG Chong, et al. Purification, characterization, and heterologous expression of a thermostable β-1,3-1,4-glucanase from Bacillus altitudinis YC-9[J]. Applied Biochemistry and Biotechnology, 2013, 169(3): 960-975.
[12] EDNEY M J, MARCHYLOB A, MACGREGOR A W. Structure of total barley β-glucan[J]. Journal of the Institute of Brewing, 1991, 97(1): 39-44.
[13] 方传记, 陆兆新, 孙力军, 等. 低能N +离子注入选育抗菌脂肽高产菌及其发酵的研究[J]. 辐射研究与辐射工艺学报, 2004, 24(6): 333-336.
[14] FANG X, YANO S, INOUE H, et al. Strain improvement of Acremonium cellulolyticus for cellulase production by mutation[J]. Journal of Bioscience and Bioengineering, 2009, 107(3): 256-261.
[15] 郭芳芳, 李金良, 陆兆新, 等. 复合诱变选育表面活性素(surfactin)高产菌株[J]. 食品科学, 2011, 32(23): 270-276.
[16] 王连峰, 陈恒雷, 张军, 等. 阿魏菇多糖高产菌株筛选的离子束诱变和复合诱变对比研究[J]. 辐射研究与辐射工艺学报, 2009, 27(4): 248-252.
[17] 万涛, 黄笛, 闻建平, 等. 石油脱硫菌Gordonia sp. WQ-01激光诱变选育及关键酶DszC克隆表达分析[J]. 化学工业与工程, 2013, 30(6): 67-73.
[18] TENG Da, WANG Jianhua, FAN Ying, et al. Cloning of β-1,3-1,4-glucanase gene from Bacillus licheniformis EGW039 (CGMCC 0635) and its expression in Escherichia coli BL21 (DE3)[J]. Applied Microbiology and Biotechnology, 2006, 72(4): 705-712.
[19] HUANG Huoqing, YANG Peilong, LUO Huiying, et al. High-level expression of a truncated 1,3-1,4-β-D-glucanase from Fibrobacter succinogenes in Pichia pastoris by optimization of codons and fermentation[J]. Applied Microbiology and Biotechnology, 2008, 78(1): 95-103.
[20] 李永仙, 谢淼, 朱林江, 等. 淀粉液化芽孢杆菌β-1,3-1,4-葡聚糖酶基因的克隆与表达[J]. 生物工程学报, 2009, 25(4): 542-548.
Breeding of High-Yield β-1,3-1,4-Glucanase-Producing Strain by Mutagenesis, and Cloning and Expression of β-1,3-1,4-Glucanase Gene
CHEN Ji, GAO Peng, LU Zhaoxin, LÜ Fengxia, ZHANG Chong, ZHAO Haizhen, BIE Xiaomei*
(College of Food Science and Technology, Nanjing Agricultural University, Nanjing 210095, China)
Abstract:In order to obtain a high-yield β-1,3-1,4-glucanase producing strain, Bacillus altitudinis YC-9 was used as the starting strain and mutagenized sequentially with nitrosoguanidine (NTG) and N +beam. The preliminary and secondary screening results showed that strain N-2-2 and 10-30s-3 had high ability to produce β-1,3-1,4-glucanase, with a yield of 28.6 and 36.1 U/mL, which revealed a 2.36- and 2.98-fold increase compared with that of the original strain, respectively, while exhibiting reduced growth rates. Furthermore, the β-1,3-1,4-glucanase gene was successfully cloned and expressed in E. coli. The intracellular activity was 79.2 U/mL, a 6.5-fold increase compared with the original strain, when induced at 30 ℃ for 6 h.
Key words:Bacillus altitudinis; β-1,3-1,4-glucanase; nitrosoguanidine (NTG) mutagenesis; N +beam implantation; heterologous expression
中图分类号:TS201.3
文献标志码:A
文章编号:1002-6630(2015)01-0179-06
doi:10.7506/spkx1002-6630-201501034
收稿日期:2014-03-14
作者简介:陈计(1988—),男,硕士研究生,研究方向为食品生物技术。E-mail:2011108007@njau.edu.cn
*通信作者:别小妹(1964—),女,教授,博士,研究方向为食品微生物及生物技术。E-mail:bxm43@njau.edu.cn