曹 琳 1,朱 莉 2,詹晓北 1,*,郑志永 1,吴剑荣 1
(1.江南大学生物工程学院,糖化学与生物技术教育部重点实验室,江苏 无锡 214122;2.江苏瑞光生物科技有限公司,江苏 无锡 214125)
摘 要:建立高效液相色谱分别测定结冷胶中乙酰基和甘油酰基含量的方法。通过水解方法将酰基从结冷胶分子中游离出来,酸化为乙酸和甘油酸(或酸根离子),醇沉将结冷胶除去,高效液相色谱检测有机酸的含量,再根据有机酸与相应酰基的分子质量关系,用内标法,并以丙酸作为内标物,计算得到2 种酰基的含量。使用HPX-87H色谱柱,以5 mmol/L硫酸溶液作为流动相,在柱温35 ℃,流速0.6 mL/min条件下,利用紫外检测器进行检测,检测波长210 nm。结果表明,此方法可同时定量2 种目的物,20 min内可完成测定,且灵敏度高,重复性和稳定性良好。实验测得本实验室生产的结冷胶中甘油酰基和乙酰基的质量分数分别为9.94%、3.29%,甘油酰基加标回收率为97.76%,相对标准偏差为0.59%,乙酰基加标回收率为93.49%,相对标准偏差为1.29%。
关键词:高效液相色谱法;结冷胶;乙酰基;甘油酰基
结冷胶是由伊乐藻假单胞菌(Pseudomonas elodea ATCC 31461)有氧发酵合成的微生物胞外多糖 [1],其用途非常广泛,与其他同类产品相比具有用量少、透明度高、香气释放能力强,耐酸耐热能力强等优点 [2],可作为乳化剂、悬浮剂、胶凝剂、成膜剂、润滑剂等应用于食品、石油、化工、制药中 [3]。结冷胶还具有良好的复配性,可与琼脂 [4]、明胶 [5]及葡聚糖 [6]等多糖复配,获得性质更为优良的复配胶,使其应用更为广泛。它的基本结构是由葡萄糖、鼠李糖、葡萄糖醛酸以2∶1∶1的物质的量比组成的线性四糖重复单位,分子质量为0.5×10 6D [7],具体结构如下 [8]:
→ 3)-β-D-Glcp-(1 → 4)-β-D-GlcpA-(1 →4 )-β-D-Glcp-(1 → 4)-α-L-Rhap-(1 →
商品形式的结冷胶主要有2 种,高酰基结冷胶和低酰基结冷胶 [9],目前主要集中于低酰基结冷胶的特性及应用研究 [10]。高酰基结冷胶在同一葡萄糖残基的O-2和O-6位置处分别连接有甘油酰基和乙酰基,每个重复单元约有1 个甘油酰基和0.5 个乙酰基 [11]。低酰基结冷胶是由天然结冷胶在热碱溶液中脱酰基后得到的 [12],酰基含量不同,结冷胶形成的凝胶性质也不同,高酰基结冷胶凝胶柔软、弹性、黏着力强;而低酰基结冷胶形成的凝胶强度大,易脆裂 [13]。
很多文献对结冷胶的凝胶机制、2 种酰基对结冷胶性质的影响进行了研究。Noda等 [14]阐述了溶液中结冷胶的凝胶机制,首先,在加热条件下,结冷胶在溶液中呈无序卷曲状态,随着液体冷却,结冷胶分子逐渐转变为有序的双螺旋结构,并形成氢键及范德华力等稳定双螺旋结构的作用力。Chandrasekaran等 [15]研究了天然结冷胶上2 种酰基对双螺旋结构及结冷胶性质的影响,认为甘油酰基位于双螺旋结构内部,它改变了双螺旋的几何构型并增加双螺旋链内氢键的个数,增加了结冷胶凝胶的黏度;而乙酰基位于双螺旋结构外侧,在一定程度上阻碍双螺旋间的聚集,降低凝胶双螺旋结构的稳定性。阳离子也是影响结冷胶性质的重要因素,Huicochea等 [16]研究了离子对结冷胶凝胶温度的影响,发现加入离子会造成结冷胶凝胶温度升高,形成更为稳定有序的凝胶结构,且二价阳离子较一价阳离子更为有效。同时,Morris等 [17]认为,甘油酰基的存在会阻碍阳离子与结冷胶的连接,因此,离子对高酰基结冷胶的促凝胶作用相对较弱。
多糖中酰基含量的测定有很多方法,Mccomb等 [18]曾用比色法,利用乙酰羟肟酸和Fe 3+的显色反应测定了果胶中O-乙酰基的含量,Cheethamm等 [19]曾用高效液相色谱(high performance liquid chromatography,HPLC)方法,分别测定了黄原胶中丙酮酰基和O-乙酰基的含量,Kao等 [20]曾用高效阴离子交换色谱配电导率检测方法分别测定了微生物胞外多糖中O-乙酰基和N-乙酰基的含量。对于结冷胶上O-甘油酰基的测定研究较少,Kuo等 [21]曾将结冷胶上的甘油酰基衍生化后用气相色谱测定了其含量,秦方等 [22]曾利用酸碱滴定的方法测定了结冷胶的总酰基含量。不同厂家、批次生产的结冷胶及不同的测定方法得到的酰基含量也不尽相同。Morris等 [11]曾在其综述中报道结冷胶中的酰基含量,甘油酰基含量约10%~12%,乙酰基含量约4%,按每个四糖单元酰基个数计算,甘油酰基约0.72~0.87 个,乙酰基约0.52 个;Jay等 [23]曾报道由其实验室培养的野生型菌株生产的结冷胶,每个四糖重复单元有甘油酰基约0.49 个,乙酰基约0.6 个,Mazen等 [24]报道由其实验室生产纯化后的天然结冷胶,每个四糖重复单元有甘油酰基约为0.8 个,乙酰基约0.8 个。若能准确测得2 种酰基的含量,不仅可以更好地研究2 种酰基分别对结冷胶性质的影响,2 种酰基的总含量及比例也是监测高酰基结冷胶产品品质的重要指标之一。而酰基含量分别测定的具体方法一直少有报道,因此急需建立一种灵敏度高,准确性及重复性良好的测定方法。本实验在前人研究的基础上,用内标法定量,以丙酸作为定量内标物,考察能够分别测定甘油酰基和乙酰基含量的HPLC方法,以期为结冷胶的深入研究提供一定的实验依据。
1.1 材料与试剂
结冷胶 江南大学生化工程与生物反应器实验室 [25];一水合甘油酸半钙盐(色谱纯) 美国Sigma公司;乙酸(色谱纯) 上海沃凯化学试剂有限公司;氢氧化钠、盐酸、无水乙醇、丙酸、硫酸(均为分析纯)国药集团化学试剂有限公司;实验中所用的水均为超纯水。
1.2 仪器与设备
LC-2010A高效液相色谱仪(配有可变波长紫外检测器及LabSolutions色谱工作站) 日本岛津公司;88-1大功率磁力搅拌器 常州国华电器有限公司。
1.3 方法
1.3.1 结冷胶样品水分和灰分含量的测定
结冷胶样品中水分含量测定按照GB 5009.3—2010《食品中水分的测定》中直接干燥方法进行 [26];灰分含量测定按照GB 5009.4—2010《食品中灰分的测定》中马弗炉灼烧的方法进行 [27],分别进行3 次平行实验。按下式计算:
式中:X 1为结冷胶中水分或灰分的含量;m 1为烘干前称量瓶和结冷胶的总质量或灼烧后坩埚和剩余灰分总质量/g;m 2为烘干后称量瓶和结冷胶的总质量或坩埚质量/g;m 0为初始称量的结冷胶样品质量/g。
1.3.2 混合标准溶液的配制
以乙酸和甘油酸作为标准物,以丙酸作为定量内标物。精确量取3 种物质适量,用水定容,配制成质量浓度0.75 g/L甘油酸标准溶液,质量浓度0.45 g/L乙酸标准溶液,质量浓度1.05 g/L丙酸标准溶液。将3 种标准溶液等体积混合,制成混合标准溶液。再配制质量浓度0.70 mg/L的丙酸溶液以备后用。将配制好的标准溶液置于4 ℃冰箱中保存。进行HPLC检测前,过0.22 μm滤膜。
1.3.3 结冷胶样品预处理
取适量的结冷胶样品,加入去离子水100 mL在80 ℃条件下充分搅拌溶解,配制成质量浓度为0.50 g/100 mL的均匀溶胶。取1 mL该溶胶于50 mL离心管中,加入1 mL 0.20 mol/L NaOH溶液,混匀后放于80 ℃水浴摇床中水解30 min,摇床转速150 r/min,将乙酰基和甘油酰基从结冷胶上完全脱落下来。
取出上述溶液,冷却至40~50 ℃时,加入6 mL乙醇,剧烈振荡混匀,放置4 ℃醇沉1~1.5 h,将离心管置于离心机中8 000 r/min,离心30 min后,结冷胶形成贴壁紧实的沉淀,取上清液6 mL转移至另一10 mL离心管,将结冷胶大分子和游离酰基溶液分开。将离心管置于真空干燥箱内,70 ℃干燥至白色固体,加入0.30 mol/L盐酸调节pH值至酸性,混匀并用水定容至1 mL,同时加入质量浓度0.70 g/L丙酸标准溶液1 mL作为定量内标物。过0.22 μm的微滤膜,进样,每个样品重复进样3 次。
1.3.4 HPLC检测条件
液相条件1:Aminex HPX-87H色谱柱(4.6 mm× 250 mm,5 μm);柱温35 ℃;流动相:5 mmol/L硫酸等度洗脱;流速0.6 mL/min;紫外检测器检测,波长210 nm;进样量20 μL。
液相条件2:Diamonsil C 18(2)色谱柱(4.6 mm× 250 mm,5 μm);柱温30 ℃;流动相体积比为甲醇(A)∶0.05%磷酸溶液(B)=3∶97,等度洗脱;流速0.8 mL/min;紫外检测器检测,波长210 nm;进样量20 μL。
1.3.5 比色法和滴定法测定酰基含量
实验中利用秦方等 [22]报道的滴定法、陈玲 [28]和罗秉俊 [29]等所报道的比色法测定结冷胶样品中的酰基含量,并将结果与本实验所述的HPLC方法测定结果进行比较。
2.1 结冷胶样品水分和灰分含量测定
发酵提取后的结冷胶样品中仍然含有水分和少量无机离子等灰分,要对结冷胶样品进行成分分析,了解其中多糖的实际含量,才能准确得到酰基在结冷胶中所占质量分数。
通过干燥法和灼烧法分析结果可知,在结冷胶样品中,水分含量为9.53%,灰分含量为5.26%,样品中多糖的实际含量为85.22%。
2.2 色谱条件的选择
乙酰基和甘油酰基与结冷胶主链上的羟基连接,形成酯键。在样品预处理过程中,通过NaOH溶液水解将乙酰基和甘油酰基游离下来,成为R—COONa羧酸盐形式,加入盐酸将其酸化为有机酸(即乙酸和甘油酸),再进行HPLC检测,即可得到2 种有机酸的质量浓度。2 种酰基的含量可根据有机酸与相应酰基的分子质量关系计算得到。
HPLC检测条件的选择,主要从测定波长、色谱柱以及内标物质等方面进行考察。选择合适的色谱柱和色谱条件,才能将2 种物质目的峰有效分开,同时选择合适的内标物质,才能对目的物准确定量。
2.2.1 测定波长的选择
根据有机酸羧基的紫外光吸收特性,对目的物溶液在波长200~300 nm处进行全波长扫描,发现在波长210 nm处,3 种目的物均有较强吸收峰,因此选取210 nm作为检测波长。
2.2.2 色谱柱的选择
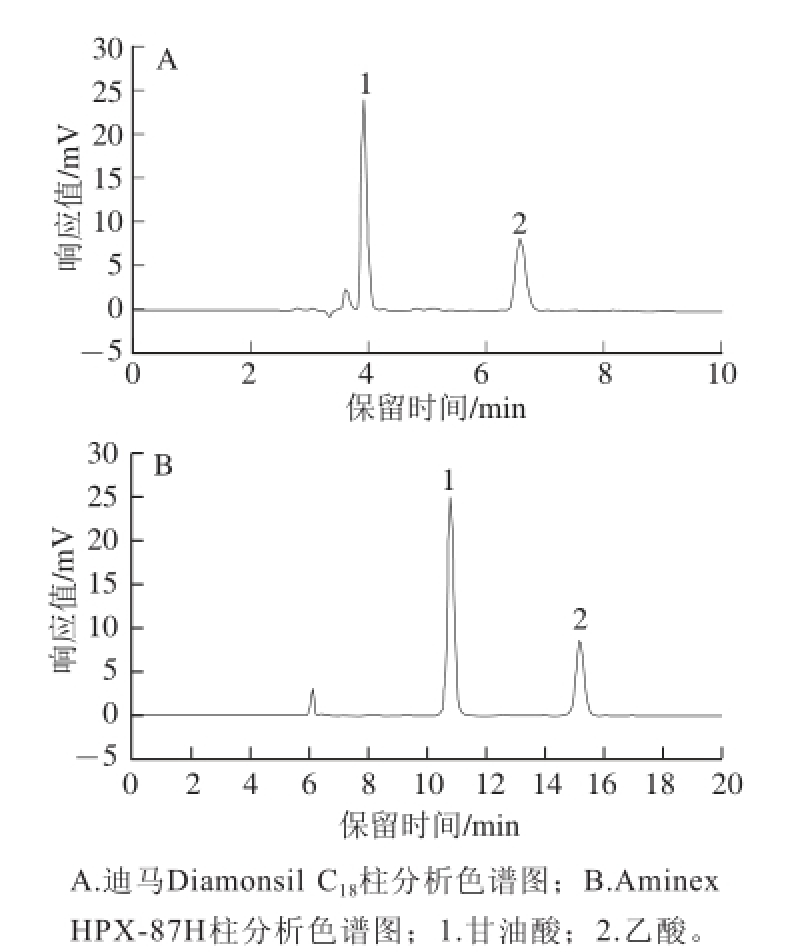
图1 不同色谱柱分析甘油酸和乙酸标准溶液色谱图
Fig.1 Chromatogram of glyceric acid and acetic acid standards separated by different columns
实验中选用迪马Diamonsil C 18(2)和Bio Rad Aminex HPX-87H 2 种色谱柱,观察2 种目的物在2 个色谱柱上的分离效果。选用C 18色谱柱可以将标准品中2 种目的物分开,10 min内即可全部检测,如图1A所示,但甘油酸出峰时间过早(3.912 min),在样品测定中受到溶剂峰和杂峰的严重干扰,乙酸目的峰也受到杂峰干扰,不能准确定量。且流动相中甲醇含量过低,仅为体积分数3%,若增加甲醇的含量,甘油酸出峰更早,对检测无益;降低甲醇含量,有机相含量过低会影响色谱柱的使用寿命。改变流速和柱温,目的峰受到杂峰干扰的情况没有改善。
HPX-87H色谱柱通过离子调节分配层析技术,根据待分析样品的多种化学特性分离混合物。实验中用稀硫酸作流动相,可以将标准溶液中2 种有机酸有效分开,如图1B所示,甘油酸出峰时间10.803 min,乙酸出峰时间15.183 min,20 min内即可将2 种物质全部洗脱下来,可以看到2 个目的峰分离度较好,且在样品色谱图中不受杂峰干扰,可以准确定量。因此本实验中优先选用Bio-Rad Aminex HPX-87H色谱柱。
2.2.3 内标物质的选择
定量内标物应是原样品中不存在的物质,性质与待测组分相近,不参与反应,必须与样品中目的物出峰时间不重合。实验中选取柠檬酸、苹果酸、酒石酸、乳酸和丙酸进行HPLC检测,几种酸出峰时间分别为8.106、9.891、8.657、12.621 min和18.007 min,由于样品经过碱水解反应处理,杂峰较多,柠檬酸、酒石酸和乳酸出峰处都受杂峰干扰;普通苹果酸存在同分异构体,出现2 个峰,无法定量;而丙酸出峰处无杂峰干扰,出峰时间与乙酸接近且不重叠,可以准确定量,因此,选用丙酸作为内标物质。
2.3 方法考察实验
确定色谱检测条件后,通过对该方法检出限和定量限、线性关系及范围、重复性、稳定性和加标回收率等参数的测定,验证HPLC方法的可行性。
2.3.1 检出限、定量限及线性关系考察
将配制好的标准品溶液逐级稀释,得到一系列甘油酸和乙酸标准品溶液,按1.3.4节的HPLC方法进行检测分析。然后以目的峰峰面积为纵坐标(y),以标准品物质质量浓度为横坐标(x)作图,得到线性方程和相关系数如表1所示,甘油酰基在12.80~358.64 mg/L范围内线性关系良好,乙酰基在7.33~220.04 mg/L范围内线性关系良好。
将标准溶液进一步稀释并检测,以信噪比为3时的标准品质量浓度作为检出限,以信噪比约为10时的标准品质量浓度作为定量限,确定检出限和定量限,结果如表1所示。
表1 甘油酰基和乙酰基回归分析、检出限和定量限
Table1 Regression analysis, limits of detection and limits of quantitation for glyceroyl and acetyll

待测物检出限/(mg/L)定量限/(mg/L)回归方程相关系数(R²)甘油酰基0.40±0.051.41±0.11y=1 714.2x+30 417.10.998 5乙酰基1.00±0.141.75±0.15y=1 740.8x+906.110.998 3
2.3.2 重复性实验
按照1.3.3节中样品处理方法,平行制备6 份结冷胶溶胶样品,按照1.3.4节中的HPLC方法进行检测,记录峰面积,并按照峰面积计算目的物含量。结果测得甘油酰基质量浓度为(159.16±2.52)mg/L,平行实验的相对标准偏差(relative standard deviation,RSD)为1.10%,乙酰基质量浓度为(53.22±3.61)mg/L,平行实验的RSD为4.04%,精密度良好。
2.3.3 稳定性实验
取预处理后的样品溶液,按1.3.4节中的HPLC方法,分别在0、4、8、12、16、20、24 h进样,按照峰面积计算目的物含量,并计算其RSD。表2结果显示,甘油酰基质量浓度的RSD为0.12%,乙酰基质量浓度的RSD为0.21%,可见试品溶液在24 h内稳定。
表2 甘油酰基和乙酰基稳定性实验
Table2 Stability for glyceroyl and acetyl

待测物含量/(mg/L)RSD/% 0 h4 h8 h12 h16 h20 h24 h甘油酰基159.92160.19159.86160.12160.09159.98159.610.12乙酰基55.8955.8555.6555.9955.7055.7655.800.21
2.3.4 加标回收率实验
配制0.50 g/100 mL的结冷胶溶胶,取1 mL样品共7 份,其中1 份作为对照溶液,直接按照1.3.3节中的方法进行处理,其余6 份在处理前,每份中加入质量浓度59.34 g/L甘油酸标准品10 μL,以及15.02 g/L乙酸标准品10 μL,然后按照1.3.3节中的方法进行样品前处理,进样检测,并按照峰面积计算目的物含量。
表3 甘油酰基和乙酰基加标回收率
Table3 Recoveries for glyceroyl and acetyl

待测目的物实际测得质量浓度/(mg/L)加入标准溶液质量浓度/(mg/L)加标回收率/%RSD/%甘油酰基184.72±1.81188.9497.76±0.950.59乙酰基38.56±0.9541.2593.49±2.331.29
测定结果如表3所示,其中甘油酰基的加标回收率平均值为97.76%,精密度RSD为0.59%,乙酰基的加标回收率平均值为93.49%,精密度RSD为1.29%,可知方法准确性良好。
2.4 结冷胶产品酰基含量测定

图2 混合标准溶液和结冷胶样品色谱图
Fig.2 Chromatograms of mixed standards and gellan gum sample
按照1.3.3节对结冷胶样品进行处理,并进行HPLC检测。混合标准溶液和结冷胶样品色谱图如图2所示。酰基含量测定结果如表4所示。
表4 结冷胶样品酰基含量测定
Table 4 Determination of acyl contents in gellan gum sample

待测目的物质量分数/%总酰基质量分数/%每个四糖重复单元中个数甘油酰基9.94±0.1513.24±0.150.83±0.01乙酰基3.29±0.130.57±0.02
由表4可知,本实验室生产的结冷胶甘油酰基和乙酰基质量分数分别为9.94%、3.29%,总酰基质量分数为13.24%。在结冷胶每个四糖重复单元中所占个数为甘油酰基0.83、乙酰基0.57。由图2B可以看到,甘油酸和乙酸目的峰分离较好,虽然样品色谱图中杂峰较多,但没有影响目的峰,丙酸出峰时间18.007 min,峰形良好,与目的峰有较好的分离度,且不受杂峰干扰,因此HPLC方法可以准确定量,且能够分别测定2 种酰基的质量浓度。
2.5 HPLC方法与其他测定方法的比较
实验中利用滴定法和比色法测定相同结冷胶样品的酰基含量,这2 种方法只能测定总酰基含量,滴定法测得结冷胶总酰基质量分数为(13.96±2.21)%,比色法测得结冷胶总酰基质量分数为(14.59±0.78)%。HPLC法测定结果如表4所示,2 种方法的测定结果与HPLC方法相比皆偏大。
滴定法原理为碱性条件下发生酯化反应逆反应并消耗碱 [11],再用酸滴定得到消耗的碱的量,滴定时利用酚酞做指示剂,实验操作简便,使用试剂和仪器简单,但缺点在于高酰基结冷胶在60 ℃以下则形成凝胶,如果在室温条件下用酸滴定,酸不能均匀分布于溶胶中,造成滴定终点时需要酸的体积偏大,测定结果偏大;如果在加热状态下滴定,会影响酚酞和碱的显色平衡反应向生成酚酞二钠盐(红色)方向移动,因此到达滴定终点所需酸体积偏大,滴定结果也偏大,且由数据可以看出结果波动性较大,重复性差。在HPLC方法和比色法的样品前处理过程中,将结冷胶与游离酰基溶液有效分开,且不产生影响测定的副产物,是准确测定酰基含量的关键步骤。在HPLC方法检测中,如果结冷胶凝胶进入色谱柱就很难将其冲洗出来,凝胶的存在会堵塞色谱柱并滋生细菌,对色谱柱造成永久性损害;在比色法中利用羟肟酸和Fe 3+的显色反应测定酰基含量 [18],显色反应需在室温条件下进行,同时加入Fe 3+,进一步促进了结冷胶凝胶的形成,测定吸光度时发生丁达尔效应,造成测定结果偏大。且凝胶双螺旋结构中可能包埋一定量的酰基,若在显色物形成后离心除去结冷胶,会损失大量显色复合物,造成结果偏小。若按本实验所述样品前处理方法,得到游离酰基溶液再进行比色测定,由于样品需经过碱水解处理,产生其他副产物,且加入的碱溶液改变了显色溶液的pH值,也会造成比色结果不可信。同时,滴定法和比色法都只能测定总酰基含量,而HPLC方法可以同时测定2 种酰基各 自的含量,这对于深入研究2 种酰基各自对结冷胶性质的影响具有重要意义。
本研究建立了检测结冷胶中2 种酰基含量的HPLC方法。由于2 种酰基(O-甘油酰基和O-乙酰基)的性质较为相近,常用的滴定法和比色法都只能测定总酰基含量,且准确性和可行性还有待考证。本实验考察的HPLC法能分别测定结冷胶上2 种酰基的含量,预处理过程中将胶体和待测物质分开,有效避免了大分子物质对色谱柱的不良影响,内标法定量降低误差,结果更为准确,方法灵敏度高,稳定性和重复性良好,分析效率高。酰基含量测定,是初步分析结冷胶结构和性质的基础,可促进研究不同酰基含量对结冷胶性质产生差异的分子结构原因。同时,酰基含量是高酰基结冷胶产品品质的重要评价指标,本方法可准确分析高酰基结冷胶产品中不同酰基组成差异,有利于建立更高标准的产品检测方法和生产规范,同时有利于开发具有精确结构特征的结冷胶生产工艺。
参考文献:
[1] 詹晓北, 王卫平, 朱莉. 食用胶的生产、性能与应用[M]. 北京: 中国轻工业出版社, 2003: 20-35.
[2] 刘丽娜, 穆莎茉莉, 贾洪峰. 新型微生物多糖结冷胶的特性及其应用研究[J]. 四川食品与发酵, 2006, 42(1): 28-31.
[3] 文一, 赵国华. 一种新型微生物胞外多糖: 结冷胶[J]. 中国食品添加剂, 2003(3): 49-52.
[4] BANERJEE S, BHATTACHARYA S. Compressive textural attributes opacity, and syneresis of gels prepared from gellan, agar and their mixtures[J]. Journal of Food Engineering, 2011, 102(3): 287-292.
[5] 王锴, 胡序建, 黄明, 等. NaCl存在条件下结冷胶以及结冷胶/明胶共混凝胶质构和熔点的研究[J]. 食品工业科技, 2013, 34(2): 108-111; 116.
[6] AHMAD N H, AHMED J, HASHIM D M, et al. Oscillatory and steady shear rheology of gellan/dextran blends[J]. Journal of Food Science and Technology, 2014: 1-8.
[7] 詹晓北. 结冷胶[J]. 中国食品添加剂, 1999(2): 66-69.
[8] JANSSON P E, LINDBERG B, SANDFORD P A. Structural studies of gellan gum, an extracellular polysaccharide elaborated by Pseudomonas elodea[J]. Carbohydrate Research, 1983, 124(1): 135-139.
[9] 聂凌鸿, 彭华松. 微生物胞外多糖-结冷胶的生产与应用前景[J]. 生命的化学, 2002, 22(2): 178-181.
[10] 孟岳成, 邱蓉. 高酰基结冷胶(HA)特性的研究进展[J]. 中国食品添加剂, 2008(5): 45-49.
[11] MORRIS E R, NISHINARI K, RINAUDO M. Gelation of gellan: a review[J]. Food Hydrocolloids, 2012, 28(2): 373-411.
[12] 张晨, 谈俊, 朱莉, 等. 糖醇对结冷胶凝胶质构的影响[J]. 食品科学,2014, 35(9): 48-52. doi: 10.7506/spkx1002-6630-201409011.
[13] 文春溪, 刘钟栋. 结冷胶研究进展[J]. 中国食品添加剂, 2014(1):204-207.
[14] NODA S, FUNAMI T, NAKAUMA M, et al. Molecular structures of gellan gum imaged with atomic force microscopy in relation to the rheological behavior in aqueous systems. 1. Gellan gum with various acyl contents in the presence and absence of potassium[J]. Food Hydrocolloids, 2008, 22(6): 1148-1159.
[15] CHANDRASEKARAN R, RADHA A, THAILAMBAL V G. Roles of potassium ions, acetyl and L-glyceryl groups in native gellan double helix: an X-ray study[J]. Carbohydrate Research, 1992, 224: 1-17.
[16] HUICOCHEA E F, HERNANDEZ A I R, SOLARES T E, et al. Sol-gel transition temperatures of high acyl gellan with monovalent and divalent cations from rheological measurements[J]. Food Hydrocolloids, 2013, 31(2): 299-305.
[17] MORRIS E R, GOTHARD M G E, HEMBER M W N, et al. Conformational and rheological transitions of welan, rhamsan and acylated gellan[J]. Carbohydrate Polymers, 1996, 30(2): 165-175.
[18] MCCOMB E A, MCCREADY R M. Determination of acetyl in pectin and in acetylated carbohydrate polymers[J]. Analytical Chemistry,1957, 29(5): 819-821.
[19] CHEETHAM N W H, PUNRUCKVONG A. An HPLC method for the determination of acetyl and pyruvyl groups in polysaccharides[J]. Carbohydrate Polymers, 1985, 5(6): 399-406.
[20] KAO G, TSAI C M. Quantification of O-acetyl, N-acetyl and phosphate groups and determination of the extent of O-acetylation in bacterial vaccine polysaccharides by high-performance anion-exchange chromatography with conductivity detection (HPAEC-CD)[J]. Vaccine,2004, 22(3): 335-344.
[21] KUO M S, MORT A J, DELL A. Identification and location of L-glycerate, an unusual acyl substituent in gellan gum[J]. Carbohydrate Research, 1986, 156: 173-187.
[22] 秦方, 詹晓北, 朱莉, 等. Gellan Gum(激冷胶)中酰基含量的测定[J].食品与机械, 1997, 13(5): 32-33.
[23] JAY A J, COLQUHOUN I J, RIDOUT M J, et al. Analysis of structure and function of gellans with different substitution patterns[J]. Carbohydrate Polymers, 1998, 35(3): 179-188.
[24] MAZEN F, MILAS M, RINAUDO M. Conformational transition of native and modified gellan[J]. International Journal of Biological Macromolecules, 1999, 26(2): 109-118.
[25] 詹晓北, 朱莉, 朱天海. 新型微生物多糖胶联多糖[J]. 工业微生物,1996, 26(2): 27-34; 45.
[26] 卫生部, 食品卫生监督检验所. GB 5009.3—2010 食品中水分的测定[S]. 北京: 中国标准出版社, 2010.
[27] 卫生部, 食品卫生监督检验所. GB 5009.4—2010 食品中灰分的测定[S]. 北京: 中国标准出版社, 2010.
[28] 陈玲, 汤丽昌, 王宁, 等. 海南库拉索芦荟的O-乙酰基含量的测定[J]. 食品研究与开发, 2011, 32(2): 103-105.
[29] 罗秉俊, 苏卫明, 刘娅. 芦荟制品中乙酰基含量测定法[J]. 中国农村科技, 2006(12): 38-39.
Determination of Glyceoyl and Acetyl in High Acyl Gellan Gum by HPLC
CAO Lin
1, ZHU Li
2, ZHAN Xiaobei
1,*, ZHENG Zhiyong
1, WU Jianrong
1
(1. Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Biotechnology,Jiangnan University, Wuxi 214122, China; 2. Jiangsu Rayguang Biotechnology Co. Ltd., Wuxi 214125, China)
Abstract:A method to measure the contents of glyceoyl and acetyl in gellan with high performance liquid chromatography(HPLC) was established. Acyl groups were dissociated from gellan by hydrolization and acidized to obtain acetic acid and glyceric acid. Then alcohol precipitation was used to remove the gel and the contents of the two kinds of organic acids were assayed by HPLC. The contents of the two acyl groups were calculated according to the corresponding relationship in molecular weight between the acyl groups and organic acids, using propionic acid as the internal standard substance. The chromatographic column used was HPX-87H with 5 mmol/L sufuric acid as the mobile phase at a fl ow rate of 0.6 mL/min. The column temperature was maintained at 35 ℃, and the analytes were detected at a UV wavelength of 210 nm. The method could quantify these two organic acids simultaneously within 20 min. It is an efficient method with excellent repeatability and stability. The contents of glyceroyl and acetyl of gellan produced in our laboratory were 9.94% and 3.29% with spiked recoveries of 97.76% and 93.49%, respectively.
Key words:high performance liquid chromatography (HPLC); gellan; acetyl; glyceroyl
中图分类号:TS227;O657.72
文献标志码:A
文章编号:1002-6630(2015)14-0059-06
doi:10.7506/spkx1002-6630-201514012
收稿日期:2014-11-05
基金项目:“十二五”国家科技支撑计划项目(2011BAD23B04);国家高技术研究发展计划(863计划)项目(2012AA021505);国家自然科学基金面上项目(31171640);无锡市科技支撑计划项目(CLE01N1208);无锡市中小企业创新基金项目(CBE01G1344)
作者简介:曹琳(1989—),女,硕士研究生,研究方向为糖生物制造。E-mail:caolin15@163.com
*通信作者:詹晓北(1962—),男,教授,博士,研究方向为工业发酵与生物化工、糖生物技术。E-mail:xbzhan@yahoo.com