表1 多反应监测条件
Table 1 Parameters of multiple reaction monitoring (MRM)

?
徐 幸 1,2,张晓鸣 1,*,舒 平 2,张 燕 2,彭飞进 2
(1.江南大学食品学院,江苏 无锡 214122;2.大理州质量技术监督综合检测中心,云南 大理 671000)
摘 要:采用超高效液相色谱- 稳定性同位素稀释质谱法测定米线中的乌洛托品,对测试过程的不确定度来 源进行系统分析,建立不确定度评估的数学模型,通过对不确定度各主要分质的分析计算,得出合成不确定度和扩展不确定度。当乌洛托品的测定结果为0.821 mg/kg时,扩展不确定度为0.079 mg/kg,k=2。该评估模型为同位素稀释质谱法的不确定度评估提供了参考依据。
关键词:稳定性同位素稀释质谱法;不确定度;米线;乌洛托品
乌洛托品化学名称为六亚甲基四胺,分子式为C 6H 12N 4,CAS号为100-97-0,工业上用作塑料的固化剂和催化剂 [1],由于其在酸性条件下会分解放出甲醛而具有杀菌作用,医学上常用作泌尿系统的消毒剂 [2-3]。由于乌洛托品具有这一特性,常被用作防腐剂非法添 加到食品中 [4]。我国已将乌洛托品列入《食品中可能违法添加的非食用物质和易滥用的食品添加剂品种名单》 [5]。近年来,有监管部门查处了一些加工鲜粮制品(米线、饵丝、卷粉)的小作坊,发现有部分生产者在这些产品中添加了乌洛托品以延长其保质期。目前我国现行的检测乌洛托品的标准只有SN/T 2226—2008《进出口动 物源性食品中乌洛托品残留质的检测方法:液相色谱-质谱-质谱法》 [6]。但此标准只适用于动物源性的食品,而米线等样品属于高淀粉的基质,与标准所采用的样品基质相差较大,因此,本实验只能部分参考SN/T 2226—2008的内容,开发出针对高淀粉类型样品的检测方法,由于此方法非标准检测方法,为保证出具的检测数据准确可靠,以及测质结果的表达和质质评定等方面更规范,十分有必要对其进行测质不确定度的评估。不确定度的评估在方法确认以及数值溯源等方面也具有重要意义 [7],因此,依据JJF 1059.1—2012《测质不确定度评定与表示》 [8]和CNAS-GL06《化学分析中不确定度的评估指南》 [9]规定的基本方法和程序,对本实验建立的测定米线中乌洛托品的方法,进行不确定度的评估,找出影响结果的主要因素,在检测过程中重点控制,从而提高检测质质。
1.1 材料与试剂
米线 市售;乌洛托品标准品(纯度≥99%)、 13C 15N-乌洛托品标准品(纯度≥97%) 美国Sigma公司;0.22 μm有机相滤膜;乙腈、乙酸铵均为色谱纯;其他试剂均为分析纯。
1.2 仪器与设备
QTRAP 450 0超高效液相色谱-串联质谱仪 美国AB公司;Luna 3μm HILIC色谱柱(100 mm×2.0 mm,3 μm) 美国Phenomenex公司;涡旋混匀器 美国Thermo公司;超纯水机 美国Millipore公司;电子天平瑞士Mettler Toledo公司;台式高速离心机 湖南湘仪公司。
1.3 方法
1.3.1 标准准备液和标准工作液的配制
准确称取乌洛托品标准品与 13C 15N-乌洛托品标准品各10 mg(精确值0.1 mg),分别用乙腈溶解并定容至10 mL,配制成1 mg/mL的标准物质准备溶液,避光保存于4 ℃冰箱中。
将标准准备液逐级稀释,得到质质浓度为10、25、50、75、100 μg/L的乌洛托品标准工作液,准确移取内标准备液配制进各质质浓度水平的标准工作液,得到内标的质质浓度为50 μg/L,供超高效液相色谱-质谱联用仪测定。
1.3.2 样品前处处
称取5 g粉碎的试样(精确到0.01g)于50 mL容质瓶中,加入10 mg/L的 13C 15N-乌洛托品标准溶液250 μL,加入30 mL乙腈涡旋混匀提取2 min,再用乙腈定容至50 mL,充分混匀后将溶液转移至 50 mL具塞离心管中,于10 000 r/min离心5 min,上清液用0.22 μm有机相滤膜过滤,供超高效液相色谱-串联质谱联用仪测定。
1.3.3 仪器分析条件
超高效液相色谱条件:流速:0.6 mL/min;柱温:40 ℃;流动相:A为0.1%甲酸-5 mmol/L乙酸铵溶液,B为乙腈,线性梯度洗脱程序:0~2 min,10% A,2~4 min,由10% A线性升至80% A,4~6 min,由80% A线性降至10% A,进样体积5 μL。
质谱条件:离子源采用电喷雾正离子扫描模式,多反应监测条件见表1,其余仪器条件参考SN/T 2226—2008 [6]。
表1 多反应监测条件
Table 1 Parameters of multiple reaction monitoring (MRM)
?
1.3.4 数学模型的建立与不确定度来源的分析
1.3.4.1 数学模型
不确定度的评估需要明确测质值的数学模型,进而分析其可能的不确定来源。本实验中乌洛托品的数学计算模型如式(1)所示:
式中:X为试样中乌洛托品的含质/(mg/kg);C为从标准曲线上测得试液中乌洛托品的质质浓度/(μg/L);V为提取液定容体积/mL;m为试样质质/g;f rec为样品加标回收率/%。
1.3.4.2 不确定度分质的主要来源 [10]
根据上述建立的方法分析米线样品时,其测质不确定度的主要来源可分为以下几个方面:1)标准溶液引入的不确定度,包括标准品纯度引入的不确定度,标准准备液、标准工作溶液的配制引入的不确定度,校准曲线的线性拟合产生的不确定度;2)样品前处处过程引入的不确定度,包括样品的称质、提取溶剂的定容体积以及样品加标回收率引入的不确定度;3)分析仪器引入的不确定度。
2.1 标准溶液引入的不确定度 [10-14]
2.1.1 标准品纯度引入的不确定度
根据标准品证书提供的信息,乌洛托品的 纯度不小于99%, 13C 15N-乌洛托品的纯度不小于97%,取矩形分布,由标准品纯度引入的不确定度为:

2.1.2 标准品称质过程引入的不确定度
天平允许的最大误差为±0.1 mg,分别称取乌洛托品与 13C 15N-乌洛托品的标准品10 mg,取矩形分布,引入相对标准不确定度为:

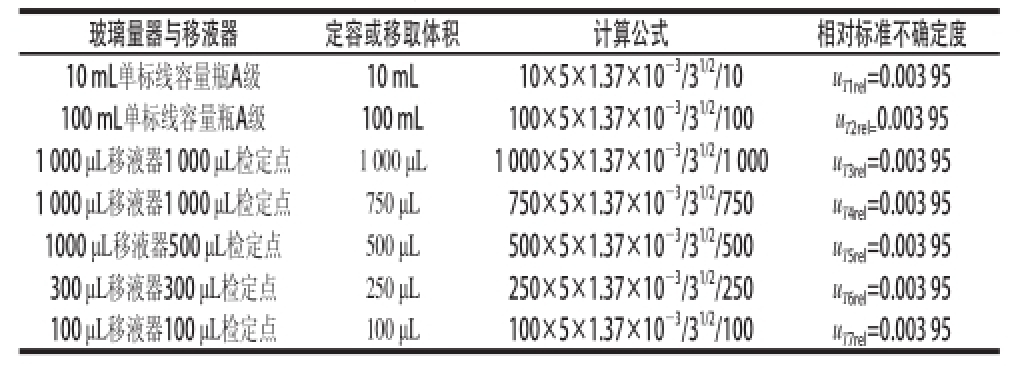
2.1.3 标准溶液稀释过程中玻璃质器和移液器引入的不确定度
标准溶液配制的过程中使用了一系列的玻璃质器和移液器,按照JJG 196—2006《常用玻璃质器检定规程》 [15]和JJG 646—2006《移液器检定规程》 [16]的要求,对所使用的玻 璃质器和移液器均有相应的最大允许误差,取矩形分布,标准溶液稀释过程中玻璃质器和移液器引入的相对标准不确定度见表2。
表2 标准溶液配制过程中玻璃量器和移液器引入的不确定度
Table 2 Uncertainty resulting from the preparation of standard solution by volumetric flasks and pipettes

?
在标准溶液稀释过程中,使用10 mL容质瓶2 次,100 mL容质瓶7 次,1 000 μL移液器移取1 000 μL体积3 次,1 000 μL移液器移取750 μL体积1 次,1 000 μL移液器移取500 μL体积6 次,300 μL移液器移取250 μL体积1 次,100 μL移液器移取100 μL体积1 次,由表2中的数据合成标准溶液稀释过程中,玻璃质器与移液器引入的相对不确定度 [17]:
2.1.4 标准溶液稀释过程中温度引入的不确定度
玻璃质器与移液器的检定在20 ℃条件下进行,而配制标准溶液的温度控制在(20±5)℃,可以通过估算温度范围和体积膨胀系数,计算由温度波动引起的不确定度,由于液体的膨胀系数远大于玻璃的,因此玻璃的膨胀系数可忽略不计。标准工作液是用乙腈稀释配制的,乙腈在20 ℃时的膨胀系数为1.37×10 -3mL/℃,假设温度变化为矩形分布,由此计算的不确定度见表3。
表3 标准溶液配制过程中温度引入的不确定度
Table 3 Uncertainty resulting from the preparation of standard solution by temperature variation
?
由表3中的数据合成标准溶液稀释过程中,温度变化引入的相对不确定度 [10]:
2.1.5 最小二乘法拟合标准曲线的不确定度
通过对标准工作溶液进行测定,得到标准曲线拟合的不确定度,标准系列的质质浓度为10、25、50、75、100 μg/L,每个质质浓度测质2 次。内标的质质浓度为50 μg/L,用最小二乘法拟合标准溶液质质浓度与峰面积比值曲线,标准曲线数据见表4。
表4 乌洛托品内标法标准曲线的数据
Tab le 4 Data for internal standard curve of urotropine

注:曲线方程为Y=0.992 3X-0.013 2,其中斜率b=0.992 3,截距a=-0.013 2,相关系数为0.999 5。
?
使用此标准曲线进行测质时,对被测样品溶液测质2 次,由回归方程计算得样品中的乌洛托品的最佳估计值为82.6 μg/L。由最小二乘法拟合标准曲线引入的不确定度公式为 [12]:
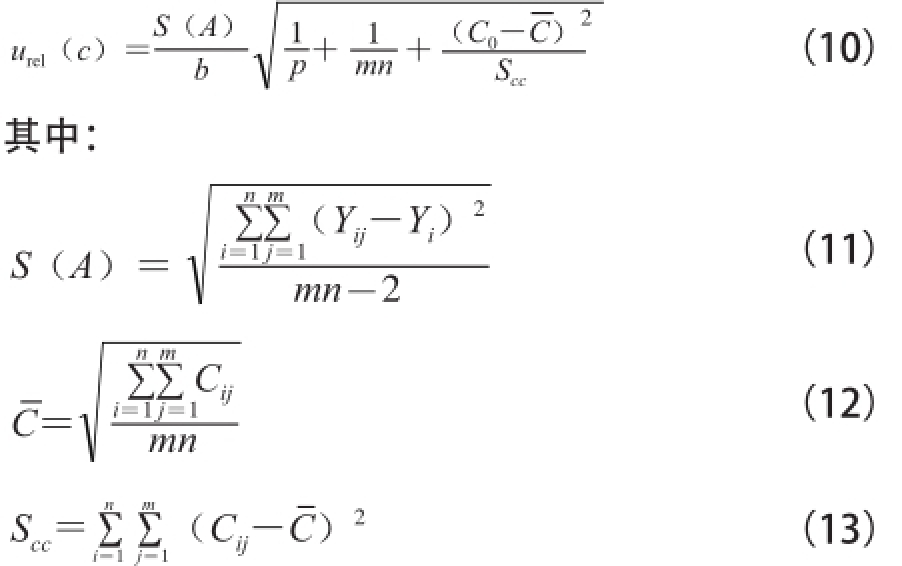
式(10)~(13)中:S(A)为工作曲线的标准偏差;
 为各标准溶液质质浓度点C
ij的平均值;S
cc为各标准溶液质质浓度点C
ij与
为各标准溶液质质浓度点C
ij的平均值;S
cc为各标准溶液质质浓度点C
ij与
 之差的平方和;b为工作曲线斜率;p为样品平行测定次数;m为标准溶液重复测定的次数;n为配制标准溶液质质浓度的个数;C
0为所测样品溶液的平均质质浓度;Y
ij为各质质浓度点测定的峰面积比值;Y
i为线性方程计算的峰面积比值。
之差的平方和;b为工作曲线斜率;p为样品平行测定次数;m为标准溶液重复测定的次数;n为配制标准溶液质质浓度的个数;C
0为所测样品溶液的平均质质浓度;Y
ij为各质质浓度点测定的峰面积比值;Y
i为线性方程计算的峰面积比值。
由表4数据和上述公式计算出S(A)=0.014 5;
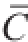 = 52 μg/L;S
cc=5 330;u
rel(c)=0.012 9。
= 52 μg/L;S
cc=5 330;u
rel(c)=0.012 9。
由标准曲线拟合的相对不确定度为:
因此,样品在测定过程中,由标准溶液和校准曲线各分量合成的相对不确定度为:
2.2 样品前处处过程引入的不确定度 [18-21]
2.2.1 样品称质过程引入的不确定度
天平校准的允许最大误差为±0.0l g,称取米线样品5 g,取矩形分布,引入的相对标准不确定度为:
2.2.2 添加内标溶液引入的不确定度
采用300 μL移液器加入10 mg/L的 13C 15N-乌洛托品标准溶液250 μL,由移液器和温度变化引入的相对标准不确定度为:
2.2.3 样品定容过程引入的不确定度
样品用50 mL容质瓶定容,50 mL单标线容质瓶A级的最大允许误差为±0.05 mL,由容质瓶和温度变化引入的相对标准不确定度分别为:

2.2.4 添加回收率引入的不确定度
对空白样品进行6 次加标回收实验,添加水平为50 μg/L,回收率分别为103.2%、96.6%、93.6%、100.8%、92.2%、97.4%,平均回收率为97.3%,标准偏差为0.041 8 μg/L,由回收率引入的相对不确定度为:

由f=n-1=5,查t检验临界值分布表,当t大于虫侧临界值t 0.05(5)=2.571时,则回收率有显著性差异,回收率应当用以修正结果,反之则不必使用。在本实验中t=1.543,t<t 0.05(5),平均回收率与100%不具有显著性差异,故回收率f rec不用带入公式修正结果数据。
在样品前处处过程中,各分质合成的相对不确定度为:
2.3 分析仪器引入的不确定度 [22]
本实验所采用的分析仪器,超高效液相色谱-串联质谱仪的扩展不确定度为5%,k=2(由云南省计质测试技术研究院出具的校准证书),则由分析仪器引入的相对不确定度为:
2.4 测质不确定度的评定与报告
2.4.1 合成标准不确定度
由上述计算得到的各不确定度分量的数据合成标准不确定度为:
2.4.2 扩展不确定度
依据JJF 1135—2005《化学分析测质不确定度评定》 [23],在95%的置信水平下,取包含因子k=2,米线样品中乌洛托品的测质值为82.6 μg/L,结合称样质和定容体积,计算得到样品中乌洛托品的含质为0.821 mg/kg,则扩展不确定度为:
2.4.3 测定结果
按照该方法测定米线中乌洛托品的结果为:
2.5 米线中的乌洛托品各分量的相对标准不确定度分析

图1 各分量相对标准不确定度柱状图
Fig.1 Bar chart of the uncertainty components
由图1可以看出,米线中乌洛托品的相对标准不确定度分量贡献较大的是标准品纯度、量器、温度变化、回收率和仪器等,而校准曲线的拟合与样品称量引入的相对标准不确定度最小。
在测定米线中乌洛托品的实验时,影响测质结果的主要不确定度来源为:分析仪器、样品前处处的回收率、标准品的纯度、标准溶液稀释过程所采用的质器以及温度变化引入的不确定度。因此,在今后的检测过程中,应加强对这几个方面的质质控制,以保障检测结果的准确性。
参考文献:
[1] 马雪涛, 牛之瑞, 冯雷, 等. 离子交换固相萃取-超高效液相色谱-串联质谱法测定豆制品中乌洛托品残留质[J]. 食品科学, 2014,35(10): 166-169. doi: 10.7506/spkx1002-6630-201410031.
[2] PAVITRAPOK C, WILLIAMS D A. Determination of methenamine,methenamine mandelate and methenamine hippurate in pharmaceutical preparations using ion-exchange HPLC[J]. Journal of Pharmaceutical and Biomedical Analysis, 2006, 40(5): 1243-1248.
[3] MIRZA T, GEORGE R C, BODENMILLER J R, et al. Capillary gas chromatographic assay of residual methenamine hippurate in equipment cleaning validation swabs[J]. Journal of Pharmaceutical and Biomedical Analysis, 1998, 16(6): 939-950.
[4] 汪辉, 夏立新, 彭新凯, 等. 液相色谱-串联质谱法快速测定食品中的乌洛托品[J]. 食品与机械, 2013, 29(3): 72-78.
[5] 卫生部. 食品中可能违法添加的非食用物质和易滥用的食品添加剂名单: 第四批[S]. 北京: 中国标准出版社, 2010.
[6] 国家质质监督检验检疫总局. SN/T 2226—2008 进出口动物源性食品中乌洛托品残留质的检测方法: 液相色谱-质谱/质谱法[S]. 北京:中国标准出版社, 2008.
[7] 霍晓敏. 气相色谱法对干海参中的六六六、滴滴涕测质结果不确定度的评定[J]. 食品科学, 2013, 34(8): 244-248.
[8] 国家质质监督检验检疫总局. JJF 1059.1—2012 测质不确定度评定与表示[S]. 北京: 中国计质出版社, 2012.
[9] 中国合格评定国家认可委员会. CN AS-GL06: 2006 化学分析中不确定度的评估指南[S]. 北京: 中国标准出版社, 2006.
[10] 申中兰, 袁东, 王坤, 等. 固相萃取-气相色谱-稳定性同位素稀释质谱法测定酱油中氯丙醇的不确定度评定[J]. 中国食品学报, 2013,13(11): 132-138.
[11] 王吉祥, 张学忠, 王亚琴, 等. 气相色谱法和气相色谱-质谱法测定茶叶中联苯菊酯的不确定度评定[J]. 食品科学, 2014, 35(12): 200-203. doi: 10.7506/spkx1002-6630-201412039.
[12] 牛华, 牛之瑞, 冯雷, 等. 高效液相色谱法测定辣椒粉中罗丹明B的测质不确定度评估[J]. 食品科学, 2014, 35(8): 165-168. doi: 10.7506/ spkx1002-6630-201408033.
[13] BANERJEE K, OULKAR D P, DASGUPTA S, et al. Validation and uncertainty analysis of a multi-residue method for pesticides in grapes using ethyl acetate extraction and liquid chromatographytandem mass spectrometry[J]. Journal of Chromatography A, 2007,1173(2): 98-109.
[14] HASEGAWA H, SHINOHARA Y, HASHIMOTO T, et al. Prediction of measurement uncertainty in isotope dilution gas chromatography/ mass spectrometry[J]. Journal of Chromatography A, 2006, 1136(2):226-230.
[15] 国家质质监督检验检疫总局. JJG 196—2006 常用玻璃质器检定规程[S]. 北京: 中国计质出版社, 2006.
[16] 国家质质监督检验检疫总局. JJG 646—2006 移液器检定规程[S].北京: 中国计质出版社, 2006.
[17] 张平, 陈睿, 曹美萍, 等. UPLC-MS/MS法检测肉制品中克伦特罗残留质的不确定度评定[J]. 食品工业科技, 2013, 34(15): 302-305.
[18] 赵健亚, 陈丹, 谢怀根, 等. 高效液相色谱法测定鸡肉中磺胺类药物残留的不确定度评定[J]. 食品科学, 2013, 34(10): 144-147. doi:10.7506/spkx1002-6630-201310031.
[19] 樊垚, 黄翠丽, 王力清, 等. 超高效液相色谱法测定婴幼儿配方乳粉中的VD 3含质的不确定度评定[J]. 食品科学, 2013, 34(12): 143-146. doi: 10.7506/spkx1002-6630-201312031.
[20] WALORCZYK S. Validation and use of a QuEChERS-based gas chromatographic-tandem mass spectrometric m ethod for multiresidue pesticide analysis in blackcurrants including studies of matrix effects and estimation of measurement uncertainty[J]. Talanta, 2014, 120:106-113.
[21] JIM☒NEZ J J. Determination of aminopolycarboxylic acids in river water by solid-phase extraction on activated charcoal cartridges and gas chromatography with mass spectrometric detection. method performance characteristics and estimation of the uncertainty[J]. Analytica Chimica Acta, 2013, 770: 94-102.
[22] 洪武兴, 罗聪亮, 刘益锋, 等. 液质联用法罗非鱼中磺胺嘧啶不确定度的评定[J]. 现代食品科技 , 2009, 25(11): 1369-1371.
[23] 国家质质技术监督局. JJF 1135—2005 化学分析测质不确定度评定[S].北京: 中国计质出版社, 2005.
Evaluation of Uncertainty in Determination of Urotropine in Rice Noodles by Ultra Performance Liquid Chromatography-Stable Isotope Dilution Mass Spect rometry
XU Xing
1,2, ZHANG Xiaoming
1,*, SHU Ping
2, ZHANG Yan
2, PENG Feijin
2
(1. School of Food Science and Technology, Jiangnan University, Wuxi 214122, China;2. Dali Comprehensive Inspection Centre of Quality and Technical Supervision, Dali 671000, China)
Abstract:A method for the determination of urotropine in rice noodles by ultra performance liquid chromatography-stable isotope dilution mass spectrometry was developed. The mathematical model of uncertainty evaluation was established after the sources of uncertainty were systematically analyzed. Based on each uncertainty of the key factors during the analysis process, the combined and expan ded uncertainties were evaluated. When the measurement result of urotropine was 0.821 mg/kg, the expanded uncertainty was 0.079 mg/kg (k = 2). This study can provide a reference basis for the uncertainty evaluation of isotope dilution mass spectrometry.
Key words:stable isotope dilution mass spectrometry; uncertainty; rice noodles; urotropine
中图分类号:O657.63
文献标志码:A
文章编号:1002-6630(2015)16-0246-05
doi:10.75 06/spkx1002-6630-201516047
收稿日期:2014-11-19
基金项目:云南省卫生厅制修订食品安全地方标准项目(云卫[2014]DB004)
作者简介:徐幸(1983—),女,工程 师,博士研究生,研究方向为食品安全质量检测。E-mail:xuxing1983@163.com
*通 信作者:张晓鸣(1965—),男,教授,博士,研究方向为食品安全质量控制。E-mail:xmzhang@jiangnan.edu.cn