
图 1 色谱峰的确立
Fig.1 Establishment of chromatographic peaks
王 晨,李晓明,卢永翎,郑铁松,吕丽爽 *
(南京师范大学金陵女子学院,江苏 南京 210097)
摘 要:建立同时在线检测丙酮醛和乙二醛的气相色谱方法。确定检测二羰基化合物的最佳条件为:以丁二酮为内标,以邻苯二胺为衍生化试剂,邻苯二胺的用量67 倍于二羰基化合物、衍生化时间10 min、萃取溶剂二氯甲烷、超声时间15 min、萃取2 次、柱箱初始温度40 ℃、程序升温5 ℃/min,色谱柱载气流量2.0 mL/min,分流比1∶1。丙酮醛和乙二醛的定量限(R SN≈10)分别为0.06 mg/L和0.08 mg/L,检出限(R SN≈ 3)分别为0.02 mg/L和0.03 mg/L,方法灵敏度高。
关键词:丙酮醛;乙二醛;气相色谱
非酶糖基化终末产物(advanced glycation end products,AGEs)是蛋白质非酶糖基化过程产生的一系列反应的终产物,还原糖和蛋白质中的氨基酸基团发生非酶促褐变反应 [1]。正常情况下,在人体内AGEs维持在一个平衡状态,但当内源 [2-3]AGEs积累过多或者外源摄入 [4-5]过多的AGEs时,会引起阿兹海默症 [6]以及糖尿病并发症(如:糖尿病性肾病 [7]和糖尿病性视网膜疾病 [8]),从而对机体造成很大的损伤。AGEs的形成的过程中,中间产物如:丙酮醛(methylglyoxal,MGO)、乙二醛(glyoxal,GO)等可以引起蛋白质交联,形成交联性的糖基化终末产物 [9-10],破坏蛋白质的结构和功能 [11],引起组织的损伤 [12],在蛋白质糖基化过程中发挥重要作用。近年来,研究发现在一些食品中检测到二羰基化合物(dicarbonyl compounds,α-DCC),包括饮料 [13-14]、饼干 [5]、蜂蜜 [15]、咖啡 [16]和面包 [17],而这些活性α-DCC,有可能进一步诱发食品中AGEs的产生。所以,建立一套能快速、有效地检测α-DCC的方法,对于分析体内外,以及食品加工和贮藏过程中α-DCC,抑制AGEs形成具有十分重要的意义。
目前,检测α-DCC的主要方式有液相色谱法、气相色谱法(gas chromatography,GC)和电化学检测法,其中液相色谱法由于其衍生化试剂种类丰富,运行时间短,在检测α-DCC的应用中最为普遍。Degen等 [18]以喹喔啉类物质为内标,用高效液相色谱-紫外检测(high performance liquid chromatography-ultraviolet detection,HPLC-UV)法检测果汁、醋和饼干中的MGO、GO、3-葡萄 糖醛酮(3-deoxyglucosone,3-DG)、3-脱氧半乳糖(3-deoxygalactosone,3-DGal)和3-脱氧-1,2-二酮戊糖(3-deoxyp entoson,3-DPs);Gensberger等 [19]用超高效液相色谱联用二极管阵列-串联质谱(ultrahighperformance liquid chromatography with hyphenated diode array-tandem mass spectrometry,UHPLC-DAD-MS-MS)法检测软饮料中的MGO、GO、3-DG、葡糖醛酮、3-DGal、1-DG和3,4-二脱氧葡糖醛酮-3-烯。Randell等 [20]以2,3-己烷二酮和5-甲基喹喔啉作为内标,用LC-MS联用法检测小鼠血液中的MGO。Tang Dan等 [21]在研究中药材虎杖粗提物和其活性成分抑制MGO的实验中,以邻苯二胺为衍生化试剂,用5-甲基喹喔啉作为内标,在紫外波长315 nm处检测。王丽苹 [22]、Bao Mingliang [23]等在检测羰基化合物时,先将其衍生化为色谱可检测的另一种物质。本实验在检测α-DCC之前,首先要把MGO和GO衍生化为2-甲基喹喔啉和喹喔啉,才能被检测出来。文献[5,14,18,24]均以邻苯二胺为衍生化试剂,将二羰基化合物衍生化为喹喔啉类物质后,进行检测。
Khuhawar等 [25]用GC-氢火焰离子检测器(GC-hydrogen flame ion detector,GC-FID)检测MGO和GO,选择间苯二甲酸二烯丙酯(diallyl isophthalate,DAP)作为衍生化试剂,在最佳衍生化pH 3条件下,在0.09~1.04 μg/mL检测范围内,检测限达到40 ng/mL,整个检测时间设定在2.6 min,但主要存在以下2 点问题:1)反应最佳pH值为3,应用范围有限。2)反应运行时间太短,体系目标峰与杂质峰不能有效的分离,尤其是在复杂的食品体系中更复杂,影响定量测定。因此,有待进一步优化。α-DCC分子质量低,易于气化,相比于LC法,GC法运行成本较低,且FID灵敏度高,对大多数有机物都有响应。本实验拟选用GC法分析活性α-DCC的含量,建立用GC-FID同时在线检测MGO、GO的方法,检测市售10 种固体饮料中的α-DCC,为控制食品加工和贮藏过程中,有害α-DCC的测定提供理论依据。
1.1 材料与试剂
固体饮料 市售;乙酸乙酯、二氯甲烷(均为分析纯)、三氯甲烷(色谱纯) 南京化学试剂有限公司;磷酸二氢钠、磷酸氢二钠、乙醛、2,3-丁二酮、2,3-己烷二酮、3,4-己烷二酮(均为分析纯) 上海国药集团化学试剂有限公司;MGO(质量分数40%)、GO(质量分数40%)、衍生化试剂邻苯二胺(O-phenylenediamine,DB) 美国Sigma-Aldrich公司;纯净水 杭州娃哈哈集团有限公司。
1.2 仪器与设备
7820GC系统(手动进样器、柱温箱、FID检测器)美国Agilent公司;XW-80A微型漩涡混合仪 上海沪西分析仪器厂有限公司;HH-6数显恒温水浴锅 金坛市富华仪器有限公司;FA2104N电子分析天平 上海精密科学仪器有限公司;PHS-3C数字式pH计 上海三信仪表厂;KQ-300B超声波清洗器 昆山市超声仪器有限公司;HGC-12A氮气吹干仪 上海Hengao T&D公司。
1.3 方法
1.3.1 GC条件
HP-5色谱柱(30 m×0.32 mm i.d.,0.25 μm);进样温度250 ℃;压力10 psi;分流比1∶1;升温程序:初始值40 ℃,保持1 min;程序升温第1阶段4 ℃/min,升至140 ℃,保持1 min;第2阶段50 ℃/min,升至250 ℃,保持1 min;FID温度280 ℃;载气流量:H 2流量30 mL/min;空气流量300 mL/min;N 2流量25 mL/min;总运行时间30.2 min。
1.3.2 目标峰的确立
1.3.2.1 MGO和GO色谱峰的确立
根据文献[24]的测定方法加以改进,在15 mL样品管中,加入质量分数40% MGO溶液和GO溶液和纯净水总共4 mL,使MGO质量浓度分别为72、36、18、7.2 mg/L,GO质量浓度分别为58、29、14.5、5.8 mg/L,然后在样品管中加入1 mL 100 mmol/L DB,盖紧样品管盖,60 ℃水浴加热15 min,冰浴后在室温条件下加入2 mol/L乙醛1 mL,将上述溶液体系混合均匀后在60 ℃水浴加热15 min,冰浴后在室温条件下加入4 mL二氯甲烷,超声波萃取15 min,萃取3 次。氮气吹干后用0.5 mL二氯甲烷复溶,取1 μL进入GC检测。
1.3.2.2 内标峰的选择
根据文献[24]的测定方法加以改进,在15 mL样品管中,加入纯净水4 mL,然后分别加入1 mmol/L的2,3-丁二酮、2,3-己烷二酮、3,4-己烷二酮0.5 mL,具体衍生化方法同1.3.2.1节,GC进行检测。
1.3.3 衍生化条件的建立
1.3.3.1 衍生化试剂用量的选取
根据文献[24]的测定方法加以改进,在15 mL样品管中,加入1 mmol/L MGO与GO样品标准液0.5 mL,然后加入1 mmol/L 2,3-丁二酮0.5 mL,分别加入20、50、100、200 mmol/L DB 1 mL,剩余衍生化步骤同1.3.2.1节方法,GC进行检测。
1.3.3.2 衍生化时间的选取
根据文献[24]的测定方法加以改进,在15 mL样品管中,加入1 mmol/L MGO与GO样品标准液0.5 mL,然后分别加入1 mmol/L 2,3-丁二酮0.5 mL、100 mmol/L DB 1 mL,盖紧样品管盖,60 ℃水浴分别加热10、15、20、25 min,剩余衍生化步骤同1.3.2.1节方法,GC进行检测。
1.3.3.3 过量衍生物去除剂乙醛用量的确定
根据文献[24]的测定方法加以改进,在15 mL样品管中,加入1 mmol/L MGO与GO样品标准液各0.5 mL,然后分别加入1 mmol/L 2,3-丁二酮0.5 mL、100 mmol/L DB 1 mL,盖紧样品管盖,60 ℃水浴分别加热10 min,冰浴后在室温条件下分别加入0、0.5、1、2 mol/L乙醛1 mL,剩余衍生化步骤同1.3.2.1节方法,GC进行检测。
1.3.3.4 萃取溶剂的选取
根据文献[24]的测定方法加以改进,在15 mL样品管中,加入1 mmol/L MGO与GO样品标准液0.5 mL,然后分别加入1 mmol/L 2,3-丁二酮0.5 mL、100 mmol/L DB 1 mL,盖紧样品管盖,60 ℃水浴分别加热10 min,冰浴后在室温条件下分别加入2 mol/L乙醛1 mL,将上述体系混合均匀后在60 ℃水浴加热15 min,冰浴后在室温条件下分别加入2 mL二氯甲烷、三氯甲烷、乙酸乙酯溶液,超声波萃取15 min,萃取3 次。氮气吹干后分别用0.5 mL相应溶液复溶,取1 μL进入GC检测。
1.3.3.5 萃取方式的确定
根据文献[24]的测定方法加以改进,在15 mL样品管中,加入1 mmol/L MGO与GO样品标准液0.5 mL,然后分别加入1 mmol/L 2,3-丁二酮0.5 mL、100 mmol/L DB 1 mL,盖紧样品管盖,60 ℃水浴分别加热15 min,冰浴后在室温条件下分别加入2 mol/L乙醛1 mL,将上述体系混合均匀后在60 ℃水浴加热15 min,冰浴后在室温条件下分别加入2 mL二氯甲烷,分别采取超声波15 min 1、2、3 次,振荡3、5 min这5 种方式进行萃取。氮气吹干后分别用0.5 mL相应萃取溶液复溶,取1 μL进入GC检测。
1.3.4 色谱条件的确定
1.3.4.1 柱箱初始温度的选取
依据1.3.3节选出的衍生化最优条件,参照1.3.1节方法,对柱箱初始温度40、50、60、70 ℃进行考察。
1.3.4.2 升温方式的确定
依据1.3.3节选出的衍生化最优条件,在1.3.4.1节色谱条件基础上,对程序升温方式4、5、6、8 ℃/min进行考察。
1.3.4.3 色谱柱载气流量的选取
依据1.3.3节选出的衍生化最优条件,在1.3.4.2节色谱条件基础上,对色谱柱载气流量1.0、1.5、2.0、2.5 mL/min进行考察。
1.3.4.4 分流比的确定
依据1.3.3节选出的衍生化最优条件,在1.3.4.3节色谱条件基础上,对色谱柱分流比1∶1、2∶1、5∶1、1∶10和不分流进行考察。
1.3.5 方法学的考察
1.3.5.1 线性关系
取MGO和GO的对照品适量,分别精密称定,用纯净水溶解并定量稀释制成质量浓度50 mg/L的混合溶液,取2,3-丁二酮的对照品适量,用纯净水溶解并定量稀释制成质量浓度86 mg/L的溶液。精密吸取MGO与GO混合溶液0.4、1、2、4、6、8、10 mL于10 mL容量瓶中,用纯净水定容到刻度,摇匀,取出1 mL于15 mL样品管中,然后加入0.5 mL 2,3-丁二酮稀释液,依据1.3.3节选出的衍生化最优条件与1.3.4节选出的色谱学最优方法注入GC仪进行测定,计算MGO与GO和内标丁二酮的峰面积之比(R),并以峰面积之比(R)对质量浓度(C,mg/L)进行线性回归。
1.3.5.2 定量限和检出限
取MGO和GO低质量浓度对照品溶液,分别逐级稀释并进行GC测定。
1.3.5.3 精密度
在15 mL样品管中,加入1 mmol/L MGO与GO样品标准液0.5 mL,然后加入1 mmol/L 2,3-丁二酮溶液0.5 mL,依据1.3.3节选出的衍生化最优条件与1.3.4节选出的色谱学最优方法,对同一个样品连续进样6 次,取平均值,计算MGO与GO和内标丁二酮的峰面积之比,代入标准曲线中。
1.3.6 固体饮料中α-DCC的检测
精确称取市售固体饮料1 g,溶于10 mL 100 mmol/L磷酸盐缓冲溶液中,超声10 min至完全溶解,取2 mL并加入4 mL甲醇,超声10 min,然后置于-40 ℃冰箱2 h,13 000 r/min离心15 min,取上清液3 mL,加入1 mmol/L 2,3-丁二酮0.5 mL、100 mmol/L DB 1 mL,盖紧样品管盖,60 ℃水浴分别加热15 min,冰浴后在室温条件下分别加入2 mol/L乙醛溶液1 mL,将上述体系混合均匀后在60 ℃水浴加热15 min,冰浴后在室温条件下分别加入2 mL二氯甲烷溶液,分别采取超声波15 min萃取2 次。氮气吹干后分别用0.5 mL相应萃取溶液复溶,取1 μL进入GC检测。
2.1 色谱峰的确立
图 1 色谱峰的确立
Fig.1 Establishment of chromatographic peaks
在体系的内标浓度为1 mmol/L条件下,考察2,3-丁二酮为内标色谱峰,结果如图1所示。可以选择的内标物有以下几种:2,3-丁二酮、3,4-己烷二酮、2,3-己烷二酮、5-甲基喹喔啉。以邻苯二胺为衍生化试剂,采用内标参与MGO、GO共同衍生化的方式,分别形成2-甲基喹喔啉、喹喔啉。内标参与共衍生化,与单一内标物相比,可以有效地减少在衍生化过程中,由于衍生化不彻底以及操作带来的损失,使定量测定更为准确。过量的衍生化试剂DB与乙醛反应,去除干扰。
由图1可以看出,以丁二酮为内标的组在20.11 min出现比较明显的色谱峰,此色谱峰在MGO(17.50 min)、GO(15.10 min)色谱峰后,与MGO、GO保留时间差距不大,但无干扰,可见丁二酮色谱峰周围不存在其他杂质峰,干扰内标的定量测定。而2,3-己烷二酮与3,4-己烷二酮内标峰保留时间长,其色谱峰会被在29.35 min的大色谱峰包裹。29.35 min出现的大色谱峰是由过量的乙醛和内标反应形成的五元环状物质,与文献[26]报道机理一致,故选取2,3-丁二酮为内标。
2.2 衍生化条件的确定
表1 样品衍生化及预处理优化
TTaabbllee 11 OOppttiimmiizzaattiioonn ooff ccoonnddiittiioonnss ffoorr ddeerriivvaattiizzaattiioonn aanndd pretreatment of samplleess
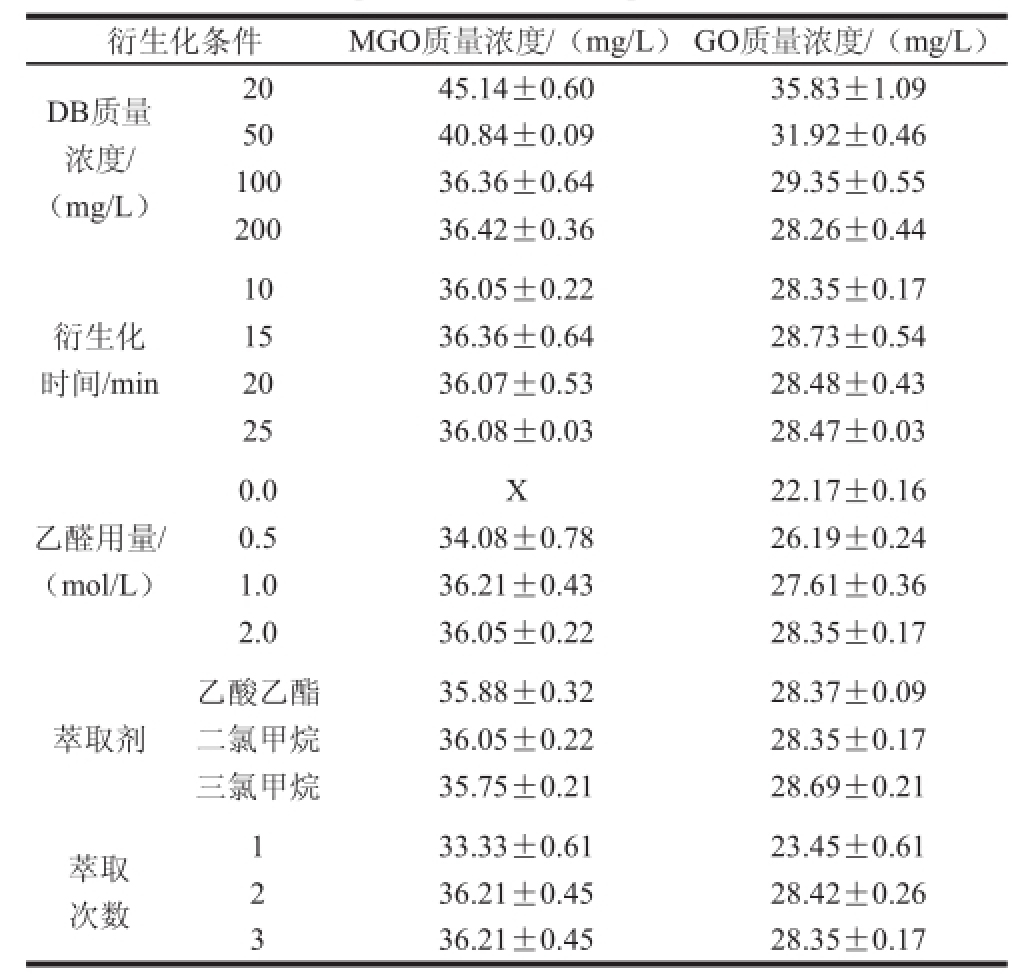
注:X.衍生化试剂DB出峰时间与MGO-DB衍生化产物相近,干扰MGO的测定。
衍生化条件 MGO质量浓度/(mg/L)GO质量浓度/(mg/L)DB质量浓度/ (mg/L)20 45.14±0.60 35.83±1.09 50 40.84±0.09 31.92±0.46 100 36.36±0.64 29.35±0.55 200 36.42±0.36 28.26±0.44衍生化时间/min 10 36.05±0.22 28.35±0.17 15 36.36±0.64 28.73±0.54 20 36.07±0.53 28.48±0.43 25 36.08±0.03 28.47±0.03乙醛用量/ (mol/L)0.0 X 22.17±0.16 0.5 34.08±0.78 26.19±0.24 1.0 36.21±0.43 27.61±0.36 2.0 36.05±0.22 28.35±0.17萃取剂乙酸乙酯 35.88±0.32 28.37±0.09二氯甲烷 36.05±0.22 28.35±0.17三氯甲烷 35.75±0.21 28.69±0.21萃取次数1 33.33±0.61 23.45±0.61 2 36.21±0.45 28.42±0.26 3 36.21±0.45 28.35±0.17
2.2.1 衍生化试剂用量
衍生化试剂的用量对于MGO、GO的衍生化过程和定量测定有着极其重要的作用,Sang Shengming等 [27]建立HPLC-DAD法考察MGO、GO与DB的衍生化反应,在37 ℃条件下,DB的加入量是MGO/GO的10~20 倍时,反应10 min,MGO和GO可以衍生化完全。Tan Di等 [24]建立GC-FID法检测饮料中的MGO,衍生化试剂DB的添加量为20 mmol/L,20 倍于α-DCC的量,反应时间15 min。本实验体系中存在乙二醛、丙酮醛、2,3-丁二酮3 种α-DCC,这3 种物质同时与DB发生衍生化反应,若衍生化试剂不足量时,3 种物质会发生竞争衍生化反应;若衍生化试剂足量甚至过量时,GO、MGO、2,3-丁二酮与DB反应完全,其测定值趋于稳定,不再发生变化,如表1所示。当衍生化试剂的量达到100 mmol/L以上,60 ℃条件下反应15 min,GC图中MGO、GO与内标的峰面积比趋于稳定。而当衍生化试剂不足量时,由于内标物未能完全衍生化,测定值忽高忽低不稳定。
2.2.2 衍生化时间的考察
衍生化时间对于MGO、GO的衍生化是否完全和定量是否准确起着较为重要的作用,当衍生化时间不足时,体系中存在的GO、MGO和2,3-丁二酮这3 种α-DCC 和DB衍生化反应的速率不同,会竞争性地与DB反应,从而引起,最终MGO和GO的测定值会有较大差异。
如表1所示,在反应10~25 min时间内,MGO与GO的值随着反应时间的延长没有发生明显变化,由此表明10 min时MGO、GO、丁二酮都已经衍生化完全,且随着反应时间的延长,MGO、GO和内标的衍生化产物很稳定。故选取衍生化时间为10 min。
2.2.3 衍生化试剂去除剂用量的确定
本实验中选择乙醛去除多余的衍生化试剂,防止过量的衍生化试剂对于色谱峰的干扰。1分子的DB可以和1~2分子的乙醛反应,形成五元环状物质。Tan Di等 [24]建立GC-FID法检测饮料中的MGO,当乙醛的量为邻苯二胺的20 倍时,乙醛能够与多余的DB反应完全。当不加入乙醛时,多余的衍生化试剂DB出峰时间与MGO-DB衍生化产物相近,完全干扰了MGO的测定。所以通过加入乙醛去除多余的DB。如表1所示,当加入乙醛浓度为2 mol/L时,能起到很好地屏蔽多余衍生化试剂的作用。
2.2.4 萃取溶剂的确定
萃取溶剂的选择主要取决于衍生化产物在有机相中的萃取效率及可操作性。乙酸乙酯、二氯甲烷和三氯甲烷3 种溶剂的极性依次为:乙酸乙酯>二氯甲烷>三氯甲烷。由表1可得,3 种衍生化试剂都能对MGO、GO、丁二酮与DB的反应产物有良好的萃取效果,萃取溶剂对MGO、GO含量的测定无显著性影响。3 种溶剂的沸点依次:乙酸乙酯77.2 ℃、二氯甲烷39.8 ℃、三氯甲烷61.3 ℃,二氯甲烷的挥发性最好,氮吹时间较短,因此选择二氯甲烷作为萃取溶剂。
2.2.5 萃取次数的考察
超声波萃取利用超声波辐射压强产生的强烈空化应效应、机械振动、扰动效应、高的加速度、乳化、扩散、击碎和搅拌作用等多级效应,增大物质分子运动频率和速度,增加溶剂穿透力,从而加速目标成分进入溶剂,促进提取的进行。旋涡振荡萃取5 min后测定的MGO质量浓度为30.08 mg/L,GO的质量浓度为22.15 mg/L,比较超声萃取的效果(萃取2 次)MGO质量浓度为36.05 mg/L,GO质量浓度为28.35 mg/L来说,萃取不完全。实验表明,超声波萃取在稳定性和萃取效果上明显高于旋涡振荡器,从表1可以得到,萃取率(萃取3 次)≈萃取率(萃取2 次)>萃取率(萃取1 次),而超声萃取2 次和3 次的效果基本一致,所以选择超声萃取2 次。
2.3 色谱条件的确定
2.3.1 初始温度的考察
表2 色谱条件的优化
Taabbllee 22 OOppttiimmiizzaattiioonn ooff cchhrroommaattooggrraapphhiicc ccoonnddiittiioonnss
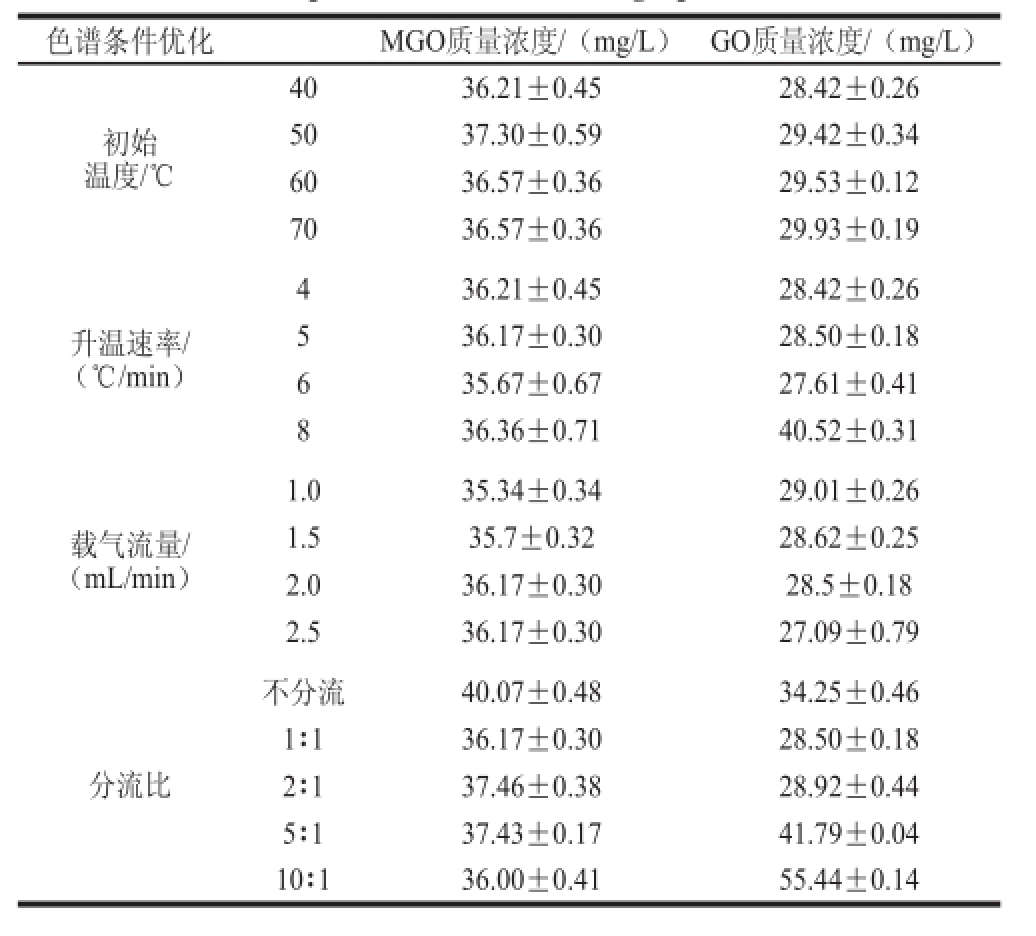
色谱条件优化 MGO质量浓度/(mg/L) GO质量浓度/(mg/L)初始温度/℃40 36.21±0.45 28.42±0.26 50 37.30±0.59 29.42±0.34 60 36.57±0.36 29.53±0.12 70 36.57±0.36 29.93±0.19升温速率/(℃/min)4 36.21±0.45 28.42±0.26 5 36.17±0.30 28.50±0.18 6 35.67±0.67 27.61±0.41 8 36.36±0.71 40.52±0.31载气流量/ (mL/min)1.0 35.34±0.34 29.01±0.26 1.5 35.7±0.32 28.62±0.25 2.0 36.17±0.30 28.5±0.18 2.5 36.17±0.30 27.09±0.79分流比不分流 40.07±0.48 34.25±0.46 1∶1 36.17±0.30 28.50±0.18 2∶1 37.46±0.38 28.92±0.44 5∶1 37.43±0.17 41.79±0.04 10∶1 36.00±0.41 55.44±0.14
柱箱初始温度的改变不影响MGO、GO、内标峰的相对保留时间差,但影响其保留时间和总运行时间。由表2可以看出,柱箱初始温度40、50、60、70 ℃时,MGO、GO的测定值基本不变,由于考虑将程序升温的初始温度升高可能在调节程序升温和流速后会产生较多的干扰杂峰,所以选择40 ℃作为柱箱初始温度。
2.3.2 程序升温方式的考察
程序升温速率的改变,不但影响MGO、GO、内标峰的保留时间和总运行时间,而且影响MGO、GO、内标峰的相对保留时间差(表2)。另外,程序升温速率4~8 ℃/min时,随着升温速率增大,目标色谱峰周围的杂质峰也越多,干扰其测定结果。所以选择5 ℃/min。
2.3.3 色谱柱载气流量的考察
色谱柱载气流速的改变,不但影响MGO、GO、内标峰的保留时间和总运行时间,而且影响MGO、GO、内标峰的相对保留时间差。由表2可以看出,色谱柱载气流速1~2.5 mL/min范围内,MGO和GO的含量没有显著性变化,提高载气流量会缩短运行时间,但随之而来的是目标色谱峰周围的杂质峰增多,干扰测定,因此选择流速2 mL/min。
2.3.4 分流比的考察
当GC进样量过大时,将考虑柱前分流问题。而分流比的大小取决于检测器的灵敏度及线性范围,出峰的大小,样品成分的响应大小。
由表2可以看出,分流比为1∶1或者2∶1时,MGO、GO的含量无显著性差别。当不分流时,MGO色谱峰和GO色谱峰会包裹其周围的基质杂质峰,引起MGO、GO的测定干扰,当分流比为5∶1或10∶1时,MGO、GO峰其出峰较小,背景杂质对其有一定的干扰,无法达到基线分离,会引起测定干扰。当不分流时,进样量大,杂质干扰较大。而当分流比超过5∶1时,样品响应值过低,无法定量GO的含量(表2)。分流比设定在1∶1~2∶1较为适合本测定方法。
2.4 方法学的考察
2.4.1 线性关系

图2 MGO(a)和GO(b)标准曲线
Fig.2 MGO and GO standard curves
由图2可知,MGO的质量浓度在0~50 mg/L范围内线性关系良好。测得MGO的线性回归方程为:R=0.017 5C+0.003 1(r=0.999 8)。GO的质量浓度在0~50 mg/L范围内线性关系良好。测得GO的线性回归方程为:R=0.017 7C+0.003 4(r=0.999 4)。
2.4.2 定量限和检测限
逐级稀释后测定的MGO和GO的定量限(R SN大于10,RSD小于2.0%)分别为0.06 mg/L(RSD=1.43%)和0.08 mg/L(RSD=1.08%),检出限(R SN≈3)分别为0.02 mg/L和0.03 mg/L。
2.4.3 精密度
表 3 精密度
Table 3 Precision of the method

化合物 含量/(mg/L) RSD/% 1 2 3 4 5 6平均MGO 35.88 35.77 35.70 35.83 36.33 36.38 35.97 0.83 GO 28.54 27.98 27.92 27.86 28.00 27.82 28.02 0.95
由表3可知,6 次平行实验得出,MGO和GO的RSD分别为0.83%、0.95%,在药典规定范围内(<2%)。表明在规定的测定条件下,精密度良好。
2.5 固体饮料中二羰基化合物的测定结果
表 4 固体饮料中α-DCC的测定结果
Taabbllee 44 DDeetteerrmmiinnaattiioonn ooff ddiiccaarrbboonnyyll ccoommppoouunnddss iinn ppoowwddeerreedd bbeevveerraaggeess μg/g

饮料编号 MGO含量 GO含量PB1 44.06±3.42 15.87±6.84 PB2 19.75±2..96 5.80±3.16 PB3 21.67±5.07 3.30±6.29 PB4 22.37±1.21 9.87±3.40 PB5 23.32±2.25 5.10±2.16 PB6 30.87±5.34 6.20±8.08 PB7 12.48±2.11 4.97±3.31 PB8 19.24±4.23 6.85±1.95 PB9 14.18±0.99 2.65±5.64 PB10 22.01±4.36 1.52±7.01
采用本实验建立的检测MGO和GO的GC方法,对市售10 种不同的固体饮料进行检测,如表4所示,其中MGO含量最高达到44.06 μg/g,GO含量最高达到15.87 μg/g,GO和MGO作为活性中间体摄入会进一步诱发AGEs的产生,由此经常饮用固体饮料会对人体带来潜在的危害。
通过对样品衍生化条件和色谱条件的优化,建立了一种同时测定MGO和GO的GC方法。具体为:以丁二酮为内标,以DB为衍生化试剂,DB用量为1 mL (100 mmol/L)、衍生化时间10 min、除去DB的乙醛用量1 mL(2 mol/L)、萃取溶剂二氯甲烷2 mL、超声萃取2 次、柱箱初始温度40 ℃、程序升温5 ℃/min、色谱柱载气流量2 mL/min,分流比1∶1。并将此方法应用于市售10 种固体饮料中二羰基化合物进行检测。表明本实验所建立的检测方法较简便、快速、成本低,对MGO和GO同时在线检测具有一定的实际意义。
参考文献:
[1] ZHANG Qibin, AMES J M, SMITH R D, et al. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: probing the pathogenesis of chronic disease[J]. Journal of Proteome Research, 2008, 8(2): 754-769.
[2] ZHANG Qibin, TANG N, SCHEPMOES A A, et al. Proteomic profi ling of nonenzymatically glycated proteins in human plasma and erythrocyte membranes[J]. Journal of Proteome Research, 2008, 7(5):2025-2032.
[3] YAMAGISHI S. Role of advanced glycation end products (AGEs)and receptor for AGEs (RAGE) in vascular damage in diabetes[J]. Experimental Gerontology, 2011, 46(4): 217-224.
[4] 吕丽爽. 牛乳加工中非酶蛋白糖基化的研究进展[J]. 食品工业科技,2013, 34(13): 359-363.
[5] ARRIBAS-LORENZO G, MORALES F J. Analysis, distribution,and dietary exposure of glyoxal and methylglyoxal in cookies and their relationship with other heat-induced contaminants[J]. Journal of Agricultural and Food Chemistry, 2010, 58(5): 2966-2972.
[6] FERRER E, ALEGRÍA A, FARRÉ R, et al. Fluorescence, browning index, and color in infant formulas during storage[J]. Journal of Agricultural and Food Chemistry, 2005, 53(12): 4911-4917.
[7] SINGH R, BARDEN A, MORI T, et al. Advanced glycation endproducts: a review[J]. Diabetologia, 2001, 44(2): 129-146.
[8] TAN A L Y, FORBES J M, COOPER M E. AGE, RAGE, and ROS in diabetic nephropathy[C]//Seminars in Nephrology. WB Saunders,2007: 130-143.
[9] BHATTACHARYYA J, SHIPOVA E V, SANTHOSHKUMAR P,et al. Effect of a single AGE modification on the structure and chaperone activity of human αB-crystallin[J]. Biochemistry, 2007,46(50): 14682-14692.
[10] 杨秀颖, 杜冠华. 糖基化终末产物及相关药物研究进展[J]. 中国药理学通报, 2011, 27(9): 1185-1188.
[11] YIM H S, KANG S O, HAH Y C, et al. Free radicals generated during the glycation reaction of amino acids by methylglyoxal[J]. Journal of Biological Chemistry, 1995, 270(47): 28228-28233.
[12] GUGLIUCCI A. Glycation as the glucose link to diabetic complications[J]. Journal American Osteopathic Association, 2000,100(10): 621-634.
[13] KALAPOS M P. Methylglyoxal in living organisms: chemistry,biochemistry, toxicology and biological implications[J]. Toxicology Letters, 1999, 110(3): 145-175.
[14] LO C Y, LI Shiming, WANG Yu, et al. Reactive dicarbonyl compounds and 5-(hydroxymethyl)-2-furfural in carbonated beverages containing high fructose corn syrup[J]. Food Chemistry, 2008, 107(3): 1099-1105.
[15] OELSCHLAEGEL S, GRUNER M, WANG P N, et al. Classifi cation and characterization of manuka honeys based on phenolic compounds and methylglyoxal[J]. Journal of Agricultural and Food Chemistry,2012, 60(29): 7229-7237.
[16] DAGLIA M, PAPETTI A, ACETI C, et al. Isolation and determination of α-dicarbonyl compounds by RP-HPLC-DAD in green and roasted coffee[J]. Journal of Agricultural and Food Chemistry, 2007, 55(22):8877-8882.
[17] URIBARRI J, WOODRUFF S, GOODMAN S, et al. Advanced glycation end products in foods and a practical guide to their reduction in the diet[J]. Journal of the American Dietetic Association, 2010,110(6): 911-916.
[18] DEGEN J, HELLWIG M, HENLE T. 1,2-Dicarbonyl compounds in commonly consumed foods[J]. Journal of Agricultural and Food Chemistry, 2012, 60(28): 7071-7079.
[19] GENSBERGER S, GLOMB M A, PISCHETSRIEDER M. Analysis of sugar degradation products with α-dicarbonyl structure in carbonated soft drinks by UHPLC-DAD-MS/MS[J]. Journal of Agricultural and Food Chemistry, 2013, 61(43): 10238-10245.
[20] RANDELL E W, VASDEV S, GILL V. Measurement of methylglyoxal in rat tissues by electrospray ionization mass spectrometry and liquid chromatography[J]. Journal of Pharmacological and Toxicological Methods, 2005, 51(2): 153-157.
[21] TANG Dan, ZHU Jiaxiao, WU Anguo, et al. Pre-column incubation followed by fast liquid chromatography analysis for rapid screening of natural methylglyoxal scavengers directly from herbal medicines:case study of Polygonum cuspidatum[J]. Journal of Chromatography A,2013, 1286: 102-110.
[22] 王丽苹, 任凤莲, 吴名剑, 等. 固相萃取毛细管气相色谱法测定卷烟滤嘴中7 种挥发性羰基化合物[J]. 分析试验室, 2009, 28(2): 116-119.
[23] BAO Mingliang, PANTANI F, GRIFFINI O, et al. Determination of carbonyl compounds in water by derivatization-solid-phase microextraction and gas chromatographic analysis[J]. Journal of Chromatography A, 1998, 809(1): 75-87.
[24] TAN Di, WANG Yu, LO C Y, et al. Methylglyoxal: its presence in beverages and potential scavengers[J]. Annals of the New York Academy of Sciences, 2008, 1126(1): 72-75.
[25] KHUHAWAR M Y, ZARDARI L A, LAGHARI A J. Capillary gas chromatographic determination of methylglyoxal from serum of diabetic patients by precolumn derivatization with 1,2-diamonopropane[J]. Journal of Chromatography B, 2008, 873(1):15-19.
[26] WANG Li, LI Chao, WANG Na, et al. Enzyme-mediated domino synthesis of 2-alkylbenzimidazoles in solvent-free system: a green route to heterocyclic compound[J]. Journal of Molecular Catalysis B:Enzymatic, 2010, 67(1): 16-20.
[27] SANG Shengming, SHAO Xi, BAI Naisheng, et al. Tea polyphenol (-)-epigallocatechin-3-gallate: a new trapping agent of reactive dicarbonyl species[J]. Chemical Research in Toxicology, 2007, 20(12):1862-1870.
Detection of Dicarbonyl Compounds in Beverages by Gas Chromatography
WANG Chen, LI Xiaoming, LU Yongling, ZHENG Tiesong, LÜ Lishuang *(Ginling College, Nanjing Normal Uni versity, Nanjing 210097, China)
Abstract:This study established an effi cient method for the detection of methylglyoxal (MGO) and glyoxal (GO) with a gas chromatograph (GC). The optimum experimental conditions were set as follows: ratio of O-phenylenediamine to α-dicarbonyl compounds, 67; derivatization time, 10 min; extraction solvent, methylene chloride; ultrasonic extraction time, 15 min; and the number of extraction, 2. The injector was operated in split mode with a split ratio of 1:1, and the carrier gas (helium)fl ow rate through the chromatographic column was 2.0 mL/min. The GC oven temperature was programmed as follows: the initial oven temperature was set as 40 ℃ and then increased at a rate of 5 ℃/min. The minimum quantitation limits (R SNapproximately equal to 10) and detection limits (R SNapproximately equal to 3) for MGO and GO were 0.06 and 0.08 mg/L,and 0.02 and 0.03 mg/L, respectively. This method was sensitive and effi cient.
Key words:methylglyoxal (MGO); glyoxal (GO); gas chromatography (GC)
中图分类号:TS201.2
文献标志码:A
文章编号:1002-6630(2015)24-0235-07
doi:10.7506/spkx1002-6630-201524044
收稿日期:2015-03-31
基金项目:江苏省基础研究计划(自然科学基金)资助项目(BK2012850)
作者简介:王晨(1991—),女,硕士研究生,研究方向为食品科学。E-mail:839541621@qq.com
*通信作者:吕丽爽(1969—),女,副教授,博士,研究方向为食品化学和功能性食品。E-mail:lishuanglv@126.com