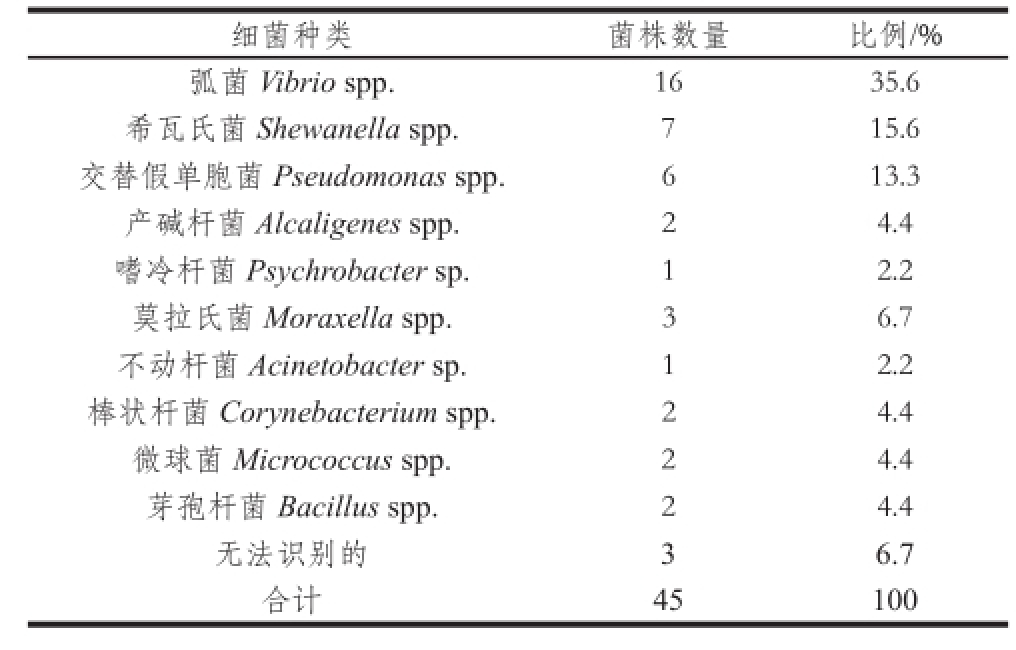
表1 基于纯培养鉴定方法的牡蛎附着细菌群落组成情况
Table 1 Microbial flora attached to oyster identified by pure culture method
?
曹 荣 1,张 井 2,孟辉辉 1,赵 玲 1,刘 淇 1,*
(1.中国水产科学研究院黄海水产研究所,山东 青岛 266071;2.温州市农业科学研究院,福建 温州 325006)
摘 要::为揭示牡蛎体附着的微生物群落结构特征,采用Illumina HiSeq高通量测序技术对牡蛎的细菌种类进行了分析,并与传统纯培养方法的分析结果进行对比。结果表明,牡蛎体附着的细菌以变形菌门(Proteobacteria,77.5%)、γ-变形菌纲(Gammaproteobacteria,72.3%)为主;在目的分类水平上,弧菌目(Vibrionales,49.3%)、交替单胞菌目(Alteromonadales,12.2%)占优势;在科的分类水平上,主要是弧菌科(Vibrionaceae,36.8%)、希瓦氏菌科(Shewanellaceae,10.3%)和交替假单胞菌科(Pseudoalteromonadaceae,7.2%);在属的分类水平上,弧菌属(Vibrio,28.3%)、希瓦氏菌属(Shewanella,10.3%)、交替假单胞菌属(Pseudoalteromonas,7.2%)比例相对较高。高通量测序分析结果中,丰度前3 位的菌属与纯培养鉴定的结果一致,但比例有所差异。
关键词:高通量测序;牡蛎;微生物群落结构
与畜禽类产品相比,水产品更易发生腐败变质,不适宜长距离物流和长时间贮存,因此鲜活水产品的消费区域受到很大的限制。水产品腐败主要是微生物作用的结果,因而鲜活水产品附着的微生物群落结构一直是国内外学者的研究热点 [1-2]。
在微生物种类鉴定技术方面,传统方法是通过富集培养、分离纯化,再根据形态、生理、生化等的实验结果得出结论。受培养条件的限制,未可培养菌通常会占到一定比例,因而传统纯培养的分析结果不能完全反映样本中微生物群落情况。随着科学技术的进步,微生物的分析手段也日趋完善,特别是以DNA为基础的分子生物学技术取得较大进展,并广泛应用到食品微生物领域 [2],如实时定量荧光聚合酶链式反应(polymerase chain reaction,PCR)技术 [3]、变性梯度凝胶电泳技术 [4]、末端限制性片段长度多态性分析技术 [5]等。近几年来,高通量测序技术快速发展,代表性的测序平台包括Illumina、Roche、Ion Torrent等,这为微生物群落分析提供了新的手段 [6]。高通量测序技术可以检测到样本中传统纯培养不能发现的低丰度细菌种类,从而更加准确、全面地反映样本的微生物群落结构。而目前该技术在水产品中的应用还较少。
牡蛎,俗称蚝、海蛎子,素有“海洋牛奶”的美称,具有很高的营养价值 [7]。贝类具有滤食的生活习性,受生长环境和摄食的影响,牡蛎体附着的微生物数量较多,种类组成也较为复杂 [8]。本研究采用Illumina HiSeq第2代测序技术对牡蛎体附着的细菌种类进行分析,并与传统纯培养方法的分析结果进行对比,研究结果既可以为鲜活水产品的微生物生态学研究策略提供参考,同时也有助于丰富牡蛎保鲜方面的基础理论。
1.1 材料
实验用牡蛎采集自青岛市胶州湾,为筏式养殖太平洋牡蛎(Crassostrea gigas)。
牡蛎采集后1 h内运至实验室,选取带壳质量为(50.0±5.0) g的个体20 只,流水冲洗后,用灭菌刀具切断闭壳肌,无菌条件下取牡蛎肉进行实验。
1.2 方法
1.2.1 平板培养和菌株种类鉴定
1.2.1.1 平板培养
配制营养琼脂基础培养基,将其中的NaCl质量浓度调整为15 g/L,制作平板备用。称取牡蛎肉约10 g,在无菌条件下匀浆,加90 mL无菌生理盐水制成10 -1稀释液,依次制备10 -2、10 -3和10 -4等稀释度。取100 μL稀释液涂布于营养琼脂平板,置于25 ℃恒温恒湿培养箱中培养48~72 h。
1.2.1.2 菌株种类鉴定
选取菌落数量适宜的计数平板,挑取平板上所有的菌落,划线分纯后保存在试管斜面中,以备鉴定。所有菌株做革兰氏染色实验,参照Bagge-Ravn等 [9]提出的海产品及加工环境中细菌种属鉴定方法,分别对革兰氏阳性菌和革兰氏阴性菌进行鉴定,其中氧化酶、过氧化氢酶、运动性、鞭毛、芽孢、糖发酵型、产H 2S、O/129敏感性等生理生化特征的实验方法参考文献[10]。
1.2.2 高通量测序分析方法
1.2.2.1 DNA提取和PCR扩增
采用十六烷基三甲基溴化铵法 [11]提取样本的基因组DNA,用0.8%的琼脂糖凝胶电泳检测纯度和质量浓度,稀释至1 ng/μL备用。
以稀释后的基因组DNA为模板,使用16S rDNA的V4区特异引物515F和806R进行PCR扩增。515F:5—GTTTCGGTGCCAGCMGCCGCGGTAA—3 806R:5—GCCAATGGACTACHVGGGTWTCTAAT—3
1.2.2.2 PCR产物的混样和纯化
PCR产物用2%的琼脂糖凝胶电泳进行检测。根据PCR产物质量浓度进行等量混样,混匀后用2%的琼脂糖凝胶电泳进行检测,回收目标条带。
1.2.3 文库构建与测序
使用TruSeq ®DNA PCR-Free Sample Preparation Kit试剂盒进行文库构建,构建好的文库经检测合格后,使用Hiseq 2500 PE250上机测序(由北京诺禾致源生物信息科技有限公司协助完成)。
1.2.4 测序数据处理
从下机数据中拆分出样品数据,然后截去Barcode和引物序列用于拼接,得到的拼接序列为原始数据。利用Qiime软件进行质量控制 [12],将原始数据进行截取,并过滤掉其中连续高质量碱基长度小于序列长度75%的数据。经过以上处理后得到的序列与数据库 [13]进行比对,检测并去除其中的嵌合体序列 [14],得到最终的有效数据。
1.2.5 OTU聚类和物种注释
利用Uparse软件 [13]对1.2.4节得到的有效数据进行聚类,以97%的一致性将序列聚类为OTUs(operational taxonomic units)。
采用RDP Classifier方法 [15]与GreenGene数据库 [16]对OTUs代表序列进行物种注释分析(阈值设定为0.8~1),分别在门、纲、目、科、属的分类水平上统计样本的群落组成。
2.1 传统纯培养分析结果
采用平板培养法共分离到45 株细菌,经形态学检验、生理生化鉴定,可以归为10 个属。其中弧菌属(Vibrio)、希瓦氏菌属(Shewanella)和交替假单胞菌属(Pseudoalteromonas)比例较高,分别为35.6%、15.6%和13.3%,另有6.7%的菌株未能鉴定出。这与之前研究结果 [17]以及其他学者的研究 [18]发现有所差异。鲜活水产品附着的细菌群落在很大程度上反映了其生长环境的细菌组成情况,并受生长水域、饵料、渔获季节、捕获方式等多种因素的影响 [19]。由此,有关牡蛎体附着微生物群落组成的研究结果一般差异较大。
表1 基于纯培养鉴定方法的牡蛎附着细菌群落组成情况
Table 1 Microbial flora attached to oyster identified by pure culture method
?
2.2 高通量测序分析结果
2.2.1 DNA提取与PCR扩增结果
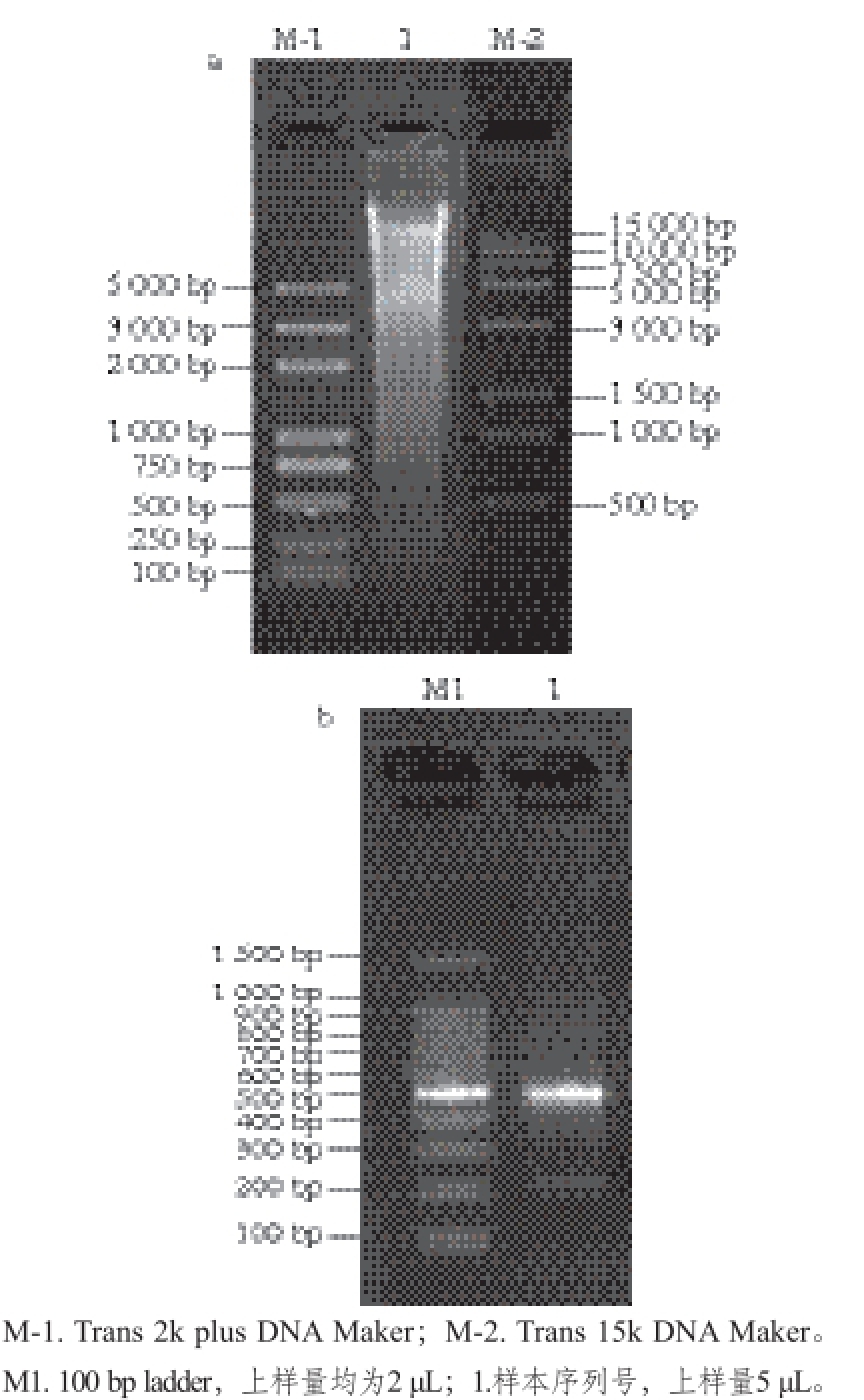
图1 提取基因组DNA(a)与PCR扩增产物(b)电泳图
Fig. 1 Electrophoretograms of genomic DNA (a) and PCR-amplified product (b)
细菌的16S rDNA具有分子大小适中、突变率低的特点,同时具有物种特异性,因此常被用作分子标签来鉴定细菌种类。提取到的细菌总DNA以及16S rDNA-V4区序列扩增的电泳图见图1。可以看出,PCR产物目标条带大小正确,总量满足2 次以上建库需要,可用于后续建库。
2.2.2 OTU物种注释与系统进化关系
采用Illumina Hiseq测序平台得到56 867 条原始数据,经拼接、过滤,得到有效序列39 390 条,其平均长度为422 bp,与16S rDNA-V4区序列长度基本一致。
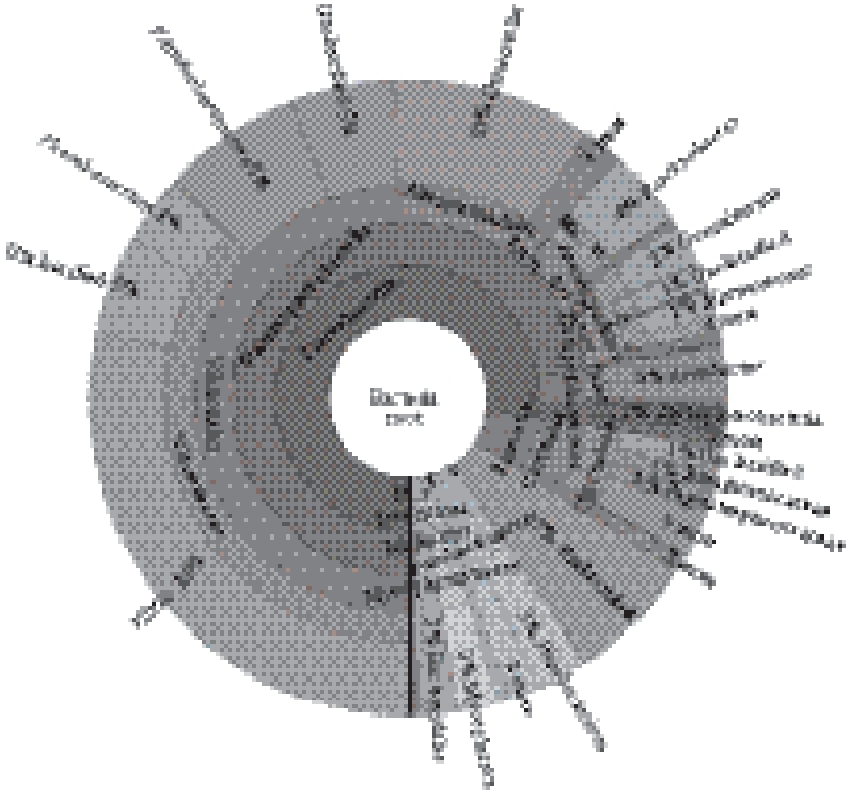
对有效数据进行聚类,得到587 个OTU。采用Krona软件 [20]对OTU进行物种注释,分析结果见图2。由内向外依次代表界、门、纲、目、科、属6 个分类级别,扇形的大小代表不同OTU注释结果的相对比例。可以看出,牡蛎中附着的细菌在变形菌门(Proteobacteria,77.5%)、厚壁菌门(Firmicutes,6.6%)、梭杆菌门(Fusobacteria,4.8%)、软壁菌门(Tenericutes,2.1%)、拟杆菌门(Bacteroidetes,2.0%)等均有分布;从纲的分类水平上来看,以γ-变形菌纲(Gammaproteobacteria,72.3%)、梭菌纲(Clostridia,5.7%)为主;在目的分类水平上,弧菌目(Vibrionales,49.3%)、交替单胞菌目(Alteromonadales,12.2%)、梭菌目(Clostridiales,5.7%)和假单胞菌目(Pseudomonadales,4.5%)占优势;而在科的分类水平上,主要是弧菌科(Vibrionaceae,36.8%)、希瓦氏菌科(Shewanellaceae,10.3%)、交替假单胞菌科(Pseudoalteromonadaceae,7.2%)和莫拉氏菌科(Moraxellaceae,4.1%)。在属的分类水平上,弧菌属(Vibrio,28%)、希瓦氏菌属(Shewanella,10%)、交替假单胞菌属(Pseudoalteromonas,7%)、嗜冷杆菌属(Psychrobacter,4%)、发光菌属(Photobacterium,3%)比例相对较高。丰度前3 位的菌属与纯培养鉴定的结果一致,但比例有所差异。纯培养方法对细菌的活性要求较高,且限于培养条件,只有一部分细菌可被培养,因此纯培养方法检测出的细菌种类相对较少,存在较大偏差的可能性也高。
通过多序列比对得到所有OTUs代表序列的系统进化关系,选取相对丰度排名前10的属所对应的OTUs数据,并结合物种注释置信度信息进行整合,结果见图3。由里往外,第1层是OTUs代表序列构建的系统发育树,分支的颜色表示其对应的属名;第2层是OTUs的相对丰度分布,柱子的高度表示OTUs的相对丰度大小;第3层是OTUs注释可信度分布,柱子的高度表示OTUs注释的可信度。可以看出,Vibrio、Shewanella和Pseudoalteromonas不仅相对丰度较高,而且在系统进化关系上也较为接近。大量研究表明,Vibrio、Shewanella和Pseudoalteromonas在水产鱼、贝类腐败过程中起到关键作用,不仅数量上占有优势,而且环境适应性较强,同时在生长繁殖过程中产生导致水产品腥臭味的小分子物质 [19]。
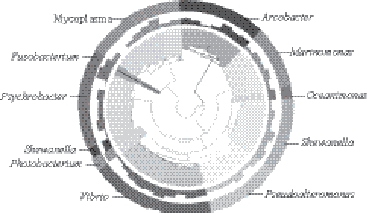
图3 OTUs的系统发育关系
Fig. 3 Phylogenetic relationship of OTUs
2.2.3 样品复杂度分析
稀释曲线和系列统计学分析指数可以评估样品中微生物群落的物种丰富度和多样性 [21]。其中,稀释曲线是从样品中随机抽取一定测序量的数据,统计它们所代表OTUs数目,以抽取的测序数据量与对应的OTUs数来构建曲线。稀释曲线可直接反映测序数据量的合理性,并间接反映样品中物种的丰富程度。由图4可以看出,随着测序数量的增加,稀释曲线趋向平坦,说明测序数据量渐进合理,更多的数据量只会产生少量新的物种(OTUs)。Chao1指数(587.2)和Shannon指数(5.48)与稀释曲线一致,说明测序数据量足够大,可以反映样品中绝大多数的微生物信息。
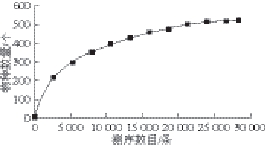
图4 稀释曲线
Fig. 4 Rarefaction curve
基于DNA的分子生物学技术已成为微生物生态学研究中的重要方法,在食品微生物领域得到广泛应用。而高通量测序技术,可以一次性对几十万至上百万条DNA分子序列进行测定,同时解决了传统微生物学基于纯培养的限制,成为微生物宏基因组学研究中的重要手段 [22]。
本实验的研究结果表明,与传统纯培养方法相比,高通量测序的研究结果更加全面地反映了牡蛎体附着微生物的物种组成及丰度信息,这为充分认识未可培养微生物并从完整的群落水平上研究微生物活动提供了可能。然而,任何一种微生物的研究手段都有其自身的优势和局限性。高通量测序技术涵盖的信息量巨大,某些未知序列因没有足够的参照标准,海量信息的归类整合就相对模糊,另外高通量测序技术是以微生物DNA作为检测基础,无法区分死菌与活菌。传统纯培养鉴定方法尽管只能进行一般的表性特征描述,但其具有成本低、分离出的菌体纯度高等特点,在水产品保鲜研究中仍占有重要地位。
从研究结果上看,在属的水平上,尽管两种方法的分析数据有所差异,但比例(丰度)前3 位的菌属均为Vibrio、Shewanella和Pseudoalteromonas。国内外学者对水产品腐败机理进行了长期研究,逐渐明确了在大多数情况下只有部分特定种类的细菌在腐败过程中占主导地位,这些导致水产品鲜度下降、品质劣化的细菌被称为特定腐败菌 [20]。Vibrio属的细菌种类多,分布广泛,在牡蛎养殖水体中 较为常见,且具有较强的分解蛋白的能力;Pseudoalteromonas在培养过程中可产生强烈的氨臭味,已被证实是多种水产鱼、贝类低温贮藏过程中的特定腐败菌 [23];而Shewanella为TMAO和H 2S产生菌,可以造成水产品贮藏过程中不良气味的产生 [24-25]。
实验发现,牡蛎体附着的优势菌集中在Vibrio、Shewanella和Pseudoalteromonas这3 个属,特别是Shewanella和Pseudoalteromonas具有较强的致腐能力,通过减菌化前处理、使用保鲜剂、改变包装方式等手段抑制这两个属细菌的生长繁殖,将有助于改善牡蛎的贮藏品质,延长货架期。
与传统纯培养方法相比,高通量测序的研究结果更加全面地反映了牡蛎体附着微生物的群落结构。在属的水平上,尽管两种方法的分析数据有所差异,但比例(丰度)前3 位的菌属均为Vibrio、Shewanella和Pseudoalteromonas。其中,Shewanella和Pseudoalteromonas对于牡蛎的贮运保鲜具有重要意义,后续研究应重点关注。
参考文献:
[1] 杨超, 车有, 宋存江. 生鲜家畜肉类与水产鱼虾类物流过程中微生物菌相变化的研究进展[J]. 食品科学, 2015, 36(23): 307-313. DOI:10.7506/spkx1002-6630-201523056.
[2] BROEKAERT K, HENDRICKX M, HERMAN L, et al. Seafood quality analysis: molecular identifi cation of dominant microbiota after ice storage on several general growth media[J]. Food Microbiology, 2011, 28(6): 1162-1169. DOI:10.1016/j.fm.2011.03.009.
[3] BJORNSDOTTIR-BUTLER K, JONES J L, BENNER R, et al. Development of a real-time PCR assay with an internal amplifi cation control for detection of Gram-negative histamine-producing bacteria in fish[J]. Food Microbiology, 2011, 28(3): 356-363. DOI:10.1016/ j.fm.2010.06.013.
[4] JIANG Y, GAO F, XU X L, et al. Changes in the composition of the bacterial fl ora on tray-packaged pork during chilled storage analyzed by PCR-DGGE and real-time PCR[J]. Journal of Food Science, 2011, 76(1): 27-33. DOI:10.1111/j.1750-3841.2010.01879.x.
[5] 任南琪, 赵阳国, 高崇洋. TRFLP在微生物群落结构与动态分析中的应用[J]. 哈尔滨工业大学学报, 2007, 39(4): 552-556. DOI:10.3321/ j.issn:0367-6234.2007.04.011.
[6] POLKA J, REBECCHI A, PISACANE V, et al. Bacterial diversity in typical Italian salami at different ripening stages as revealed by highthroughput sequencing of 16S rRNA amplicons[J]. Food Microbiology, 2015, 48(6): 342-356. DOI:10.1016/j.fm.2014.08.023.
[7] 励建荣, 徐辉. 海水双壳贝类的质量控制研究进展[J]. 食品科学, 2005, 26(增刊1): 128-134. DOI:10.3321/j.issn:1002-6630.2005. z1.034.
[8] ROTERMAN Y R, BENAYAHU Y, RESHEF L, et al. The gill microbiota of invasive and indigenous spondylus oysters from the mediterranean sea and northern red sea[J]. Environmental Microbiology Reports, 2015, 7(6): 860-867. DOI:10.1111/1758-2229.12315.
[9] BAGGE-RAVN D, NG Y, HJELM M, et al. The microbial ecology of processing equipment in different fish industries-analysis of the microflora during processing and following cleaning and disinfection[J]. International Journal of Food Microbiology, 2003, 87(3): 239-250. DOI:10.1016/S0168-1605(03)00067-9.
[10] 王秀茹. 预防医学微生物学及检验技术[M]. 北京: 人民卫生出版社, 2002: 875-880; 896-908.
[11] TURNER C R, MILLER D J, COYNE K J, et al. Improved methods for capture, extraction, and quantitative assay of environmental DNA from Asian bigheaded carp (Hypophthalmichthys spp.)[J]. PLoS One, 2014, 9(12): e114329. DOI:10.1371/journal.pone.0114329.
[12] AVERSHINA E, FRISLI T, RUDI K. De novo semi-alignment of 16S rRNA gene sequences for deep phylogenetic characterization of next generation sequencing data[J]. Microbes and Environments, 2013, 28(2): 211-216. DOI:10.1264/jsme2.ME12157.
[13] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 2010, 7(5): 335-336. DOI:10.1038/nmeth.f.303.
[14] BOKULICH N A, SUBRAMANIAN S, FAITH J J, et al. Qualityfi ltering vastly improves diversity estimates from Illumina amplicon sequencing[J]. Nature Methods, 2013, 10(1): 57-59. DOI:10.1038/ nmeth.2276.
[15] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10(10): 996-998. DOI:10.1038/nmeth.2604.
[16] EDGAR R C, HAAS B J, CLEMENTE J C, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200. DOI:10.1093/bioinformatics/btr381.
[17] CAO R, XUE C, LIU Q. Changes in microbial fl ora of Pacifi c oysters (Crassostrea gigas) during refrigerated storage and its shelf-life extension by chitosan[J]. International Journal of Food Microbiology, 2009, 131(2): 272-276. DOI:10.1016/j.ijfoodmicro.2009.03.004.
[18] ORTIGOSA M, GARAY E, PUJALTE M J. Numerical taxonomy of aerobic, Gram-negative bacteria associated with oysters and surrounding seawater of the Mediterranean coast[J]. Systematic and Applied Microbiology, 1995, 17(4): 589-600. DOI:10.1016/S0723-2020(11)80081-0.
[19] GRAM L, DALGAARD P. Fish spoilage bacteria-problems and solutions[J]. Current Opinion in Biotechnology, 2002, 13(3): 262-266. DOI:10.1016/S0958-1669(02)00309-9.
[20] LUO C, TSEMENTZI D, KYRPIDES N, et al. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample[J]. PLoS One, 2012, 7(2): 1-12. DOI:10.1371/journal.pone.0030087.
[21] CAPORASO J G, LAUBER C L, WALTERS W A, et al. Ultra-highthroughput microbial community analysis on the Illumina HiSeq and MiSeq platforms[J]. The ISME Journal, 2012, 6(8): 1621-1624. DOI:10.1038/ismej.2012.8.
[22] 聂志强, 韩玥, 郑宇, 等. 宏基因组学技术分析传统食醋发酵过程微生物多样性[J]. 食品科学, 2013, 34(15): 198-203. DOI:10.7506/ spkx1002-6630-201315041.
[23] GRAM L, HUSS H H. Microbiological spoilage of fish and fish products[J]. International Journal of Food Microbiology, 1996, 33(8): 121-137. DOI:10.1016/0168-1605(96)01134-8.
[24] DALGAARD P. Qualitative and quantitative characterization of spoilage bacteria from packed fish[J]. International Journal of Food Microbiology, 1995, 26(3): 319-333. DOI:10.1016/0168-1605(94)00137-U.
[25] GORBY Y A, YANINA S, MCLEAN J S, et al. Electrically conductive bacterial nanowires produced by Shewanella oneidensis strain MR-1 and other microorganisms[J]. Proceedings of the National Academy of Sciences, 2006, 103(30): 11358-11363. DOI:10.1073/ pnas.0604517103.
Microbial Flora Analysis of Oyster: A Comparison between Traditional Plate Culture Method and High Throughput Sequencing
CAO Rong
1, ZHANG Jing
2, MENG Huihui
1, ZHAO Ling
1, LIU Qi
1,*
(1. Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, China; 2. Wenzhou Academy of Agricultural Sciences, Wenzhou 325006, China)
Abstract:Illumina HiSeq high throughput sequencing and the traditional plate culture method were comparatively applied to investigate the structure of bacterial community attached to oyster. Results showed that the dominant bacteria attached to oyster belonged to the Gammaproteobacteria (72.3%) class in the Proteobacteria (77.5%) phylum. At the order level, Vibrionales (49.3%) and Alteromonadales (12.2%) were dominant. At the family level, Vibrionaceae (36.8%), Shewanellaceae (10.3%) and Pseudoalteromonadaceae (7.2%) were dominant. At the genus level, the relative proportions of Vibrio (28.3%), Shewanella (10.3%) and Pseudoalteromonas (7.2%) were high. The results of high throughput sequencing for the top three most dominant genera were consistent with those obtained with the traditional culture method although the proportions of these bacteria were somehow different.
Key words:high throughput sequencing; oyster; microbial fl ora
DOI:10.7506/spkx1002-6630-201624021
中图分类号:TS254.7
文献标志码:A
文章编号:
引文格式:
曹荣, 张井, 孟辉辉, 等. 高通量测序与传统纯培养方法在牡蛎微生物群落分析中的应用对比[J]. 食品科学, 2016, 37(24): 137-141.
DOI:10.7506/spkx1002-6630-201624021. http://www.spkx.net.cn
CAO Rong, ZHANG Jing, MENG Huihui, et al. Microbial flora analysis of oyster: a comparison between traditional plate culture method and high throughput sequencing[J]. Food Science, 2016, 37(24): 137-141. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201624021. http://www.spkx.net.cn
收稿日期:2016-04-11
基金项目:国家自然科学基金青年科学基金项目(31301587)
作者简介:曹荣(1981—),男,副研究员,博士,主要从事水产品保鲜研究。E-mail:caorong@ysfri.ac.cn
*通信作者:刘淇(1965—),男,研究员,本科,主要从事水产品加工与质量安全研究。E-mail:liuqi@ysfri.ac.cn