张腊梅,李道敏
*,李兆周,刘二伟,李松彪,曹 力,高红丽,王 芳,侯玉泽
(河南科技大学食品与生物工程学院,河南 洛阳 471003)
摘 要:合成功能单体N,O-双异丁烯酰乙醇胺(N,O-bismethacryloyl ethanolamine,NOBE),采用表面分子印迹技术,以硅胶为载体,乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EGDMA)为交联剂,制备乙氧酰胺苯甲酯(ethopabate,ETP)分子印迹聚合物(molecularly imprinted polymers,MIPs)。采用正交试验方法,对ETPNOBE物质的量比、ETP-EGDMA物质的量比和致孔剂种类3 个因素进行考察,结果表明:3 个影响因素主次顺序为致孔剂种类>ETP-NOBE物质的量比>ETP-EGDMA物质的量比;当致孔剂为乙腈、ETP-NOBE-EGDMA的物质的量比为1∶2∶20时,所制备的ETP-MIPs印迹效果最佳。Scatchard模型研究发现存在两类结合位点,高亲和位点与低亲和位点的平衡离解常数K d和最大表观结合量Q max分别为K d1=1.608 μg/mL,Q max1=1.101 μg/mg;K d2=0.109 μg/mL,Q max2=0.172 μg/mg。
关键词:分子印迹聚合物;表面分子印迹技术;正交试验设计;乙氧酰胺苯甲酯
乙氧酰胺苯甲酯(ethopabate,ETP),分子式为C 12H l5O 4N,相对分子质量为237.26,抗球谱广,抗球指数高,常作为一种广谱型抗球虫药物,在鸡球虫病防治方面发挥着重要的作用 [1]。然而此药物的不合理使用,不仅会使鸡产生抗药性,而且极易造成鸡肉组织中ETP的残留超标,人体食用后直接给健康带来威胁 [2]。
目前动物性食品中ETP的主要检测方法有液相色谱(liquid chromatograph,LC)法 [3-4]、高效液相色谱(high performance liquid chromatograph,HPLC)法 [5-7]、气相色谱(gas chromatography,GC)法 [4]、LC-质谱(mass spectrometry,MS)联用法 [8]和电化学传感器法 [9]等。GC及LC-MS法检测灵敏度较高,但GC法需对样品进行衍生化,操作繁琐,干扰多,适用范围有限;LC-MS法抗干扰能力强,更适用于多残留检测;而HPLC法易受到杂质干扰,对样品纯度要求高,样品前处理繁琐,且检测结果难以满足药物残留限量的要求 [10]。因此,研究出一种准确度高、灵敏度强的检测方法,用于鸡肉中ETP的控制及检测极为重要。
分子印迹技术(molecular imprinting technology,MIT)作为一种基于分子识别的分离技术倍受关注;采用该技术制备的分子印迹聚合物(molecularly imprinted polymers,MIPs)具有构效预定性、特定识别性、化学稳定性和广泛适用性等优点 [11],在食品检验、药物分离和检测等方面显示出广泛的应用前景。表面分子印迹在很大程度上改善了传统聚合方法制备的MIPs选择吸附能力低、刚性弱、传质速率慢、单分散性差和稳定性不足等缺点,近年来被广泛研究 [12]。
本研究合成了功能单体N,O-双异丁烯酰乙醇胺(N,O-bismethacryloyl ethanolamine,NOBE);并采用表面分子印迹技术,以硅胶为载体、乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EGDMA)为交联剂,制备ETP-MIPs;通过正交试验设计,从ETP-NOBE物质的量比、ETP-EGDMA物质的 量比和致孔剂种类3 个因素进行考察,对ETP-MIPs的制备条件进行了优化;并对ETP-MIPs的吸附性能进行了研究。
1.1 材料与试剂
乙醇胺(分析纯) 江苏强盛功能化学股份有限公司;三乙胺(分析纯) 天津市德恩化学试剂有限公司;甲基丙烯酰氯(纯度99%) 上海诺泰化工有限公司;柱层析硅胶(100~200、300~400目)青岛海洋化工厂;KH-570硅烷偶联剂(纯度99.5%)恒宇化工有限公司;ETP(纯度99%) 美国Sigma公司;EGDMA(纯度99%) 阿拉丁试剂公司;乙腈(色谱纯) 西陇化工股份有限公司;偶氮二异丁腈(分析纯) 通光荣化工有限公司;其他有机试剂均为国产分析纯。
1.2 仪器与设备
SZCL-3B数显控温磁力搅拌器 巩义市予华仪器责任有限公司;AC-60-S电动搅拌器 上海昂尼仪器仪表有限公司;UV2400紫外-可见分光光度计 上海舜宇恒平科学仪器有限公司;1260 HPLC仪 美国安捷伦公司;VERTEX70傅里叶变换红外光谱仪 德国布鲁克公司;STARe差示扫描量热(differential scanning calorimeter,DSC)仪 瑞士梅特勒-托利多集团;S-4800场发射扫描电镜 日本日立公司。
1.3 方法
1.3.1 功能单体的合成
参照文献[13-14]报道的方法,N 2保护及冰浴条件下,将64 mmol(3.83 mL)乙醇胺加入装有200 mL CH 2Cl 2的500 mL四口瓶中,以400 r/min的速率进行机械搅拌,然后向混合溶液中缓慢加入148 mmol(20.6 mL)三乙胺,搅拌15 min后,逐滴缓慢加入148 mmol(14.4 mL)甲基丙烯酰氯,将温度缓慢升至室温后反应24 h。反应结束后,对所得产物进行抽滤,弃除滤渣,滤液依次用30 mL 5 mol/L的NaHCO 3溶液和30 mL 5 mol/L的柠檬酸钠溶液进行洗涤,收集淡黄色有机层液体,加入10 g无水Na 2SO 4脱水后,过滤,滤液进行旋转蒸发,脱除有机溶剂,得到黄色油状物。采用硅胶柱层析法提纯所得产品,先用乙酸乙酯-正己烷(1∶1,V/V)混合溶液洗脱除去杂质,再用乙酸乙酯洗脱,得到目标化合物,进行旋转蒸发后,得到淡黄色油状物即为NOBE。
ν/cm -1:3 334.72[δ(N—H)],1 720.66[δ(C=O)],1 6 5 8.5 8,1 6 2 1.4 7[δ(C=C)],1 2 9 7.3 9 [δ(C—N)],1 167.82 [δ(C—O)]。 1H NMR (DMSO,400 MHz,10 -6):δ 8.08(1H,bs),6.04 (1H,m),5.64 (1H,t,J=4 Hz),5.32 (1H,s),4.14 (2H,t,J=8 Hz),3.41(2H,t,J=4 Hz),1.86(3H,s),1.84(3H,s); 13C NMR (DMSO,400 MHz,10 -6):δ 167.80,166.51,139.82,135.84,125.74,119.06,62.74,39.40,18.50,17.89。M r=197。
1.3.2 硅胶的活化与硅烷化
参考文献:[15-16]报道的方法:称取硅胶20 g加到100 mL浓硝酸中,磁力搅拌2 h,均匀后静置24 h,通过浸泡可除去无机杂质并增加其表面羟基数目。活化硅胶用去离子水反复清洗至pH 7,于100 ℃条件下干燥12 h后,备用。N 2保护条件下,250 mL三口烧瓶中加入活化硅胶以及100 mL甲苯,磁力搅拌条件下逐滴滴加KH-570硅烷偶联剂10 mL,于120 ℃条件下回流反应24 h,制得表面被不饱和C=C双健修饰的改性硅胶。将所得产物依次用甲苯、丙酮、甲醇进行清洗,于100 ℃条件下干燥12 h,得到硅烷化硅胶,装入自封袋中,封口备用。
1.3.3 ETP表面MIPs的制备
称量1 mmol ETP溶于90 mL乙腈中,加入一定量功能单体NOBE和引发剂偶氮二异丁腈(2,2’-azobis(2-methylpropionitrile),AIBN)100 mg,超声混匀后,封口,静置于冰箱中,4 ℃条件下低温预聚合12 h。准确称取2 g硅烷化硅胶加入到250 mL三口烧瓶中。向预聚合溶液中依次加入一定量交联剂EGDMA,超声混匀10 min。N 2保护条件下,将混合溶液缓慢加入四口烧瓶中,以400 r/min的转速进行机械搅拌,通N 215 min后密封,在60 ℃的水浴条件下进行聚合反应12 h。聚合反应结束后,将混合液体进行离心,弃上清液,将得到的MIPs先用超纯水进行洗涤,涡旋并离心3 次,采用同 样方法,再依次用甲醇洗涤3 次、乙醇洗涤2 次,以除去未参与聚合反应的功能单体和交联剂等其他杂质分子,最终将得到的MIPs于120 ℃条件下干燥6 h。
以甲醇-乙酸(9∶1,V/V)混合溶液为洗脱液进行洗提,洗脱液多次更换,直到紫外测试无ETP检出为止。用甲醇洗涤除去乙酸等杂质,所得MIPs于100 ℃条件下干燥,并用200~325 目分级筛进行分级,得到粒径范围在45~75 μm的MIPs。
非印迹聚合物(non-imprinted polymers,NIPs)的制备除了不加ETP外,其他与MIPs相同。
1.3.4 ETP-MIPs制备单因素试验
影响MIPs吸附效果的因素有很多,其中致孔剂的种类、功能单体用量以及交联剂用量等是其最主要的影响因素。
1.3.4.1 致孔剂种类的选择
致孔剂可使MIPs生成丰富的孔结构。对于非共价型印迹,致孔剂对MIPs的结构、形态影响较大,其极性、介电常数、黏度及用量等都会对MIPs的吸附效果产生较大影响 [17]。固定模板ETP用量1 mmol、NOBE用量4 mmol、EGDMA用量20 mmol,分别选择乙腈、氯仿、丙酮作为溶剂制备MIPs。
基于HPLC法,根据吸附前后溶液中ETP质量浓度变化计算MIPs和NIPs的吸附量Q,平行测定3 次取平均值。计算 [18]如式(1)所示:
式中:C 0为ETP的初始质量浓度/(μg/mL);C为ETP的平衡质量浓度/(μg/mL);V为溶液体积/mL;M为微球质量/mg。
印迹效果(α) [19]的计算按照公式(2)进行:
式中:Q MIPs为MIPs的吸附量/(μg/mg);Q NIPs为NIPs的吸附量/(μg/mg)。
1.3.4.2 功能单体用量的选择
功能单体为MIPs提供特定的结合位点;用量不足会引起专一识别位点少,MIPs含有较少印迹空穴;含量过多时,过量的功能单体呈随机分布状态,从而引入更多游离的功能基团,导致特异性吸附位点减少,非特异性吸附位点增多,使MIPs对模板的选择吸附能力变差 [20]。分别选择NOBE用量为2、4、6 mmol,固定模板ETP用量1 mmol、EGDMA用量20 mmol,制备ETP-MIPs。
1.3.4.3 交联剂用量的选择
交联剂可用于固定结合位点,生成与模板分子结构匹配的印迹空穴。用量较少时,所制备的MIPs交联度较低,刚性弱,比表面积较小,吸附能力降低;过量则会引起MIPs交联度过高,刚性过强,这不仅使模板分子洗脱较难,部分特异性结合位点被包埋其中,有效印迹空穴减少,导致模板分子难以与其内部的结合位点结合,MIPs吸附能力较差 [21]。分别选择EGDMA用量为10、20、30 mmol,固定模板ETP用量1 mmol、NOBE用量4 mmol,制备ETP-MIPs。
1.3.5 正交试验
为了制备出具有理想印迹效果的ETP-MIPs,采用L 9(3 4)因素水平表进行正交试验,主要从ETP-NOBE物质的量比、ETP-EGDMA物质的量比和致孔剂种类3 个因素考察(表1),在单因素试验基础上,对ETP-MIPs的制备条件进行优化。
表1 正交试验因素与水平
Table 1 The table of orthogonal experiment factor level code

水平因素A ETP-NOBE物质的量比B ETP-EGDMA物质的量比C致孔剂种类1 1∶11∶15氯仿2 1∶21∶20乙腈3 1∶31∶25氯仿-乙腈(1∶1,V/V)
1.3.6 ETP-MIPs的表征
1.3.6.1 ETP-MIPs吸附性能的测定
准确配制5 μg/mL的ETP乙腈标准溶液50 mL。准确称取各组MIPs和NIPs各20 mg,分别装入5 mL离心管中。各加入2 mL 5 μg/mL的ETP标准溶液,密封后,置于恒温摇床中,以室温25 ℃、160 r/min的振荡速率振荡6 h后,离心取上清液,0.22 μm有机滤膜过滤,进行HPLC检测。
1.3.6.2 傅里叶红外光谱检测
对普通硅胶、硅烷化硅胶、MIPs、NIPs进行红外光谱检测,采用KBr压片法,以纯KBr为底物样品,在4 000~400 cm -1范围内进行测定。通过红外图谱中一些特殊基团的特征峰进行分析,探讨ETP与NOBE之间的相互作用机理。
1.3.6.3 DSC检测
分别称取10~20 mg MIPs和NIPs,在N 2环境下,50~350 ℃温度范围内(升温速率5 ℃/min )进行测试及分析,以评价ETP-MIPs和ETP-NIPs的热稳定性。
1.3.6.4 扫描电镜检测
通过场发射扫描电镜测试,放大500~10 000倍后,观察ETP-MIPs、ETP-NIPs的微观结构,研究其结构及形貌。
1.3.6.5 等温吸附曲线的测定与Scatchard分析
准确配制质量浓度为0.1、0.2、0.5 、1、1.5、2、3、4、5、10、15、20 μg/mL的系列ETP乙腈标准工作液。分别称取ETP-MIPs、ETP-NIPs各20 mg,装入一系列5 mL离心管中。各离心管加入2 mL不同质量浓度的ETP标准溶液,密封后,置于恒温摇床中,以室温25 ℃、160 r/min的速率振荡6 h,取出并进行离心,取上清液1 mL经0.22 μm的有机滤膜过滤,用HPLC检测。根据吸附前后标准溶液的质量浓度改变,计算不同质量浓度水平条件下ETP-MIPs和ETP-NIPs的结合量。以ETP标准溶液的初始质量浓度为横坐标,以单位质量ETPMIPs、ETP-NIPs对ETP的结合量Q为纵坐标,绘制等温吸附曲线。
根据Scatchard模型 [22]对ETP-MIPs的结合性能进行分析研究,其方程如式(3)所示:
式中:Q为单位质量ETP-MIPs对ETP的结合量/(μg/mg);C为达到吸附平衡后ETP的质量浓度/(μg/mL);Q max为最大表观结合量/(μg/mg);K d为结合位点的平衡离解常数/(μg/mL)。
以单位质量ETP-MIPs对ETP的结合量Q为横坐标,以Q/C为纵坐标作图,进行线性拟合,并求得结合位点的K d与Q max。
2.1 ETP-MIPs制备条件的优化
2.1.1 单因素试验结果
2.1.1.1 致孔剂种类的选择
表2 致孔剂种类对MIPs印迹效果的影响
Table 2 Influence of different porogens on the adsorption efficiency of MIPs
致孔剂种类乙腈氯仿丙酮印迹效果2.3281.869 0.506
如表2所示,致孔剂为乙腈时,MIPs印迹效果最好,选择乙腈做致孔剂制备MIPs。
2.1.1.2 功能单体用量的选择
表3 功能单体用量对MIPs印迹效果的影响
Table 3 Influence of functional monomer dosage on the adsorption efficiency of MIPs
NOBE用量/mmol246印迹效果2.6271.4350.786
如表3所示,NOBE用量为2 mmol时,ETP-MIPs印迹效果最好,而NOBE用量为6 mmol时ETP-MIPs印迹效果最差,这是由于过量的功能单体使得ETP-MIPs特异吸附能力降低。因此选择ETP与NOBE物质的量比为1∶2制备MIPs。
2.1.1.3 交联剂用量的选择如表4所示,当EGDMA用量为20 mmol时,ETPMIPs印迹效果最好;用量为10、20 mmol时,印迹效果均不理想,这是由于交联剂用量不足或过量都会引起MIPs
表4 交联剂用量对MIPs印迹效果的影响
Table 4 Influence of crosslinking agent dosage on the adsorption efficiency of MIPs
EGDMA用量/mmol102030印迹效果1.2182.1570.913
中的有效印迹空穴变少,从而导致吸附能力变差。因此选择ETP与EGDMA物质的量比为1∶20制备MIPs。
2.1.2 正交试验结果
表5 ETP-MIPs制备的正交试验设计及结果
Table 5 Results of orthogonal array experiments for optimization of ETP-MIPs preparation
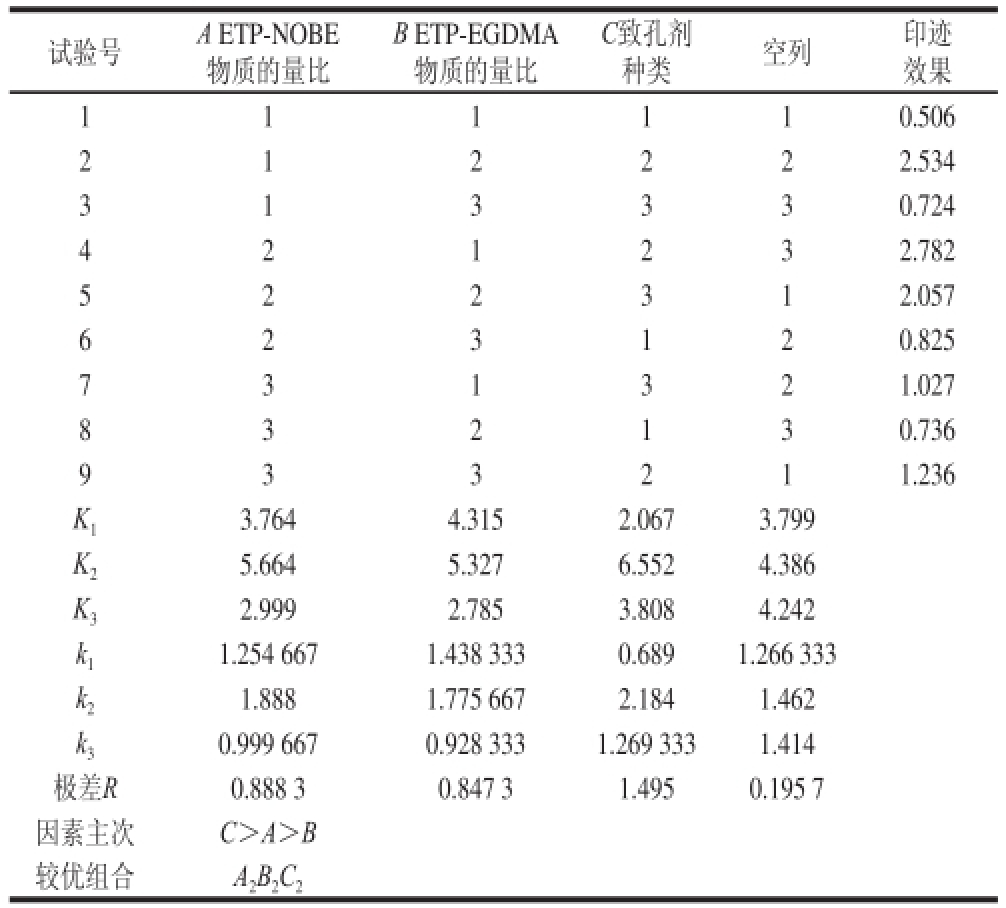
试验号A ETP-NOBE物质的量比种类空列印迹效果1 1 1 110.506 B ETP-EGDMA物质的量比C致孔剂2 222.534 3 1 3 330.724 1 2 4 232.782 5 2 2 312.057 2 1 6 120.825 7 3 1 321.027 2 3 8 130.736 9 3 3 211.236 3 2 K 13.7644.3152.0673.799 K 25.6645.3276.5524.386 K 32.9992.7853.8084.242 k 11.254 6671.438 3330.6891.266 333 k 21.8881.775 6672.1841.462 k 30.999 6670.928 3331.269 3331.414极差R0.888 30.847 31.4950.195 7因素主次C>A>B较优组合 A 2B 2C 2
ETP-MIPs制备条件的优化结果如表5所示。通过正交试验设计,对ETP-NOBE物质的量比、ETP-EGDMA物质的量比和致孔剂种类3 个因素进行考察,得到了理想MIPs的制备条件。
表5表明:3 个因素影响主次顺序为C>A>B,即致孔剂种类>ETP-NOBE物质的量比>ETP-EGDMA物质的量比;较优组合水平为A 2B 2C 2,即当致孔剂为乙腈、ETP-NOBE-EGDMA的物质的量比为1∶2∶20时,所制备的ETP-MIPs印迹效果最佳。
通过表6方差分析可知,因素A、C对指标影响显著,显著性水平为α=0.05,因素B对指标影响不显著。通过正交试验设计,可得较优组合水平为A 2B 2C 2,增加验证实验对正交结果加以确认。基于最优组合水平,即选择乙腈做致孔剂、ETP-NOBE-EGDMA的物质的量比1∶2∶20,其他条件不变,制备ETP-MIPs和ETPNIPs,通过对其吸附性能进行测试,平行测定3 次取平均值,得到ETP-MIPs的印迹效果为2.804。
表6 ETP-MIPs制备正交试验方差分析表
Table 6 Analysis of variance for optimization of ETP-MIPs preparation

注:F 0.01(2,2)=99.0,F 0.05(2,2)=19.0。
方差来源方差自由度均方差F值显著性水平A ETP-NOBE物质的量比1.255 320.627 620.115 4α=0.05 B ETP-EGDMA物质的量比1.091 920.545 917.496 8不显著C致孔剂种类3.408 421.704 254.621 8α=0.05误差0.062 420.031 2总和5.818 08
2.2 傅里叶红外光谱检测结果

图1 活化硅胶、硅烷化硅胶的红外图谱
Fig.1 Infrared spectra of activated silica gel and silylation silica gel
如图1所示,观察活化硅胶谱带,在810 cm -1附近出现Si—O—Si键的对称伸缩振动峰,450 cm -1处出现Si—O键的弯曲振动峰;1 260 cm -1处是Si—OH键的面内弯曲振动峰;在3 650 cm -1处出现的吸收峰为尖峰,且峰形比较宽,这是由于硅胶经活化后在其表面形成呈缔合状态的羟基发生伸缩振动所致。观察硅烷化硅胶谱带,接枝硅烷基后,硅胶微粒的透光率减弱,Si—O—Si骨架的峰形变宽,吸收峰强度显著增强;在950 cm -1处为一尖峰,这可能由于硅胶表面Si—OH被接枝C=C后,造成Si—O骨架局部不对称,发生伸缩振动所致;1 500 cm -1处为C=C骨架的伸缩振动峰,这表明C=C已接枝到硅胶表面;3 650 cm -1处仍存在—OH的吸收峰,说明活化硅胶表面呈缔合状态的羟基—OH未被全部接枝C=C。

图2 ETP-MIPs、ETP-NIPs的红外图谱
Fig.2 Infrared spectra of ETP-MIPs and ETP-NIPs
如图2所示,ETP-MIPs的吸收峰强度比ETP-NIPs低,ETP-MIPs的透光率略差于ETP-NIPs;ETP-MIPs和ETP-NIPs的谱带比较类似,说明二者分子结构是相似的,ETP-MIPs中的模板分子ETP已被完全洗脱干净。图谱中存在Si—O—Si键的对称伸缩振动峰和Si—O骨架的弯曲振动峰;2 920 cm -1处为烷烃—C—H的对称伸缩振动峰,主要来自NOBE、EGDMA的—CH 3和—CH 2—,以及KH-570硅烷偶联剂的—CH 2CH 3;1 260 cm -1处为—C—O—的伸缩振动峰,1 740 cm -1处出现较强的酯羰基C=O伸缩振动峰,表明ETP-MIPs、ETP-NIPs的分子结构中存在来自功能单体NOBE的基团—C—O— C=O;1 150 cm -1处为C—H键面内弯曲振动;ETP-MIPs谱带中3 400 cm -1处为分子间氢键—O—H的伸缩振动峰,对比ETP-NIPs谱带,此处ETP-MIPs的—OH吸收峰较弱,并且发生一定程度的蓝移现象,这是由于ETP-MIPs制备过程中,ETP与NOBE之间发生氢键作用,引起—OH吸收峰的强度及位移变化。
上述分析表明,ETP与NOBE之间通过氢键,在引发剂AIBN的引发聚合作用下,ETP、NOBE和EGDMA在硅胶表面发生共聚,属于非共价印迹,成功合成ETP-MIPs。
2.3 DSC检测结果

图3 ETP-MIPs、ETP-NIPs的DSC曲线
Fig.3 DSC curves of ETP-MIPs and ETP-NIPs
图3可以反映出ETP-MIPs、ETP-NIPs在50~350 ℃范围内的热稳定性及其相态转变的情况。据观察,ETPMIPs和ETP-NIPs的吸热与放热过程基本一致,相态转变相似;但ETP-NIPs出现相态转变较快,其热稳定性不如ETP-MIPs。观察ETP-MIPs曲线,50 ℃时发生吸热反应,之后达到基线平衡,这可能是由于样品干燥不完全引起的,在150~215 ℃范围内,随着温度升高,ETPMIPs发生放热反应,形成冷结晶,其玻璃化温度达到215 ℃,此后开始吸热,当温度达到280 ℃左右时,ETPMIPs开始分解。而ETP-NIPs的玻璃化温度为160 ℃,分解温度仅为225 ℃左右,其热稳定性较差。
2.4 扫描电镜检测结果

图4 ETP-MIPs、ETP-NIPs的扫描电镜图
Fig.4 Scanning electron micrographs of silicon, ETP-MIPs and ETP-NIPs
如图4所示,低倍(×500)时显示ETP-MIPs基本保持了与硅烷化硅胶类似的结构,分散性比较好,而ETPNIPs呈现出较复杂的无规则结构,团聚现象较为严重。放大到10 000倍后,可以清晰地看到硅烷化硅胶、ETPMIPs和ETP-NIPs的表面形貌,其中硅烷化硅胶表面比较均匀致密;由于多孔结构的存在,ETP-MIPs表面粗糙不平;二者图像很相似,差别不大,这主要是因为MIPs在硅胶表面的膜层很薄,与没有接枝的硅胶的区别不大。而ETP-NIPs颗粒形状不规则,粒径不均一,微粒间由于发生不规则的团聚现象导致黏连成块,分散性较差,并且表面比较光滑,凹凸感不强,没有孔穴出现,这是由于在ETP-NIPs制备过程中, 没有模板ETP参与分子印迹预组装过程而引起的。
2.5 等温吸附曲线的测定与Scatchard分析

图5 ETP-MIPs和ETP-NIPs的等温吸附曲线
Fig.5 Isotherm absorption curves of ETP-MIPs and ETP-NIPs
如图5所示,随着ETP标准溶液初始质量浓度的增大,ETP-MIPs、ETP-NIPs对ETP的结合量逐渐增多;质量浓度较高时,ETP-MIPs、ETP-NIPs实现饱和吸附,二者吸附变化趋势相同;对比相同质量浓度添加条件下的吸附量,ETP-MIPs明显高于ETP-NIPs,说明MIPs对模板分子的高度亲合力和特异识别性远大于非选择性键合作用 [23]。这可能是由于ETP-MIPs中不仅存在能与ETP分子官能团相匹配的结合位点,其表面还有能与ETP分子结构相匹配的特定形状的立体孔穴;印迹过程中,ETPMIPs对ETP具有一定的专一识别性,更有利于MIPs与ETP的结合,使得选择性吸附能力增强;而ETP-NIPs没有选择性的结合位点,对ETP的吸附主要依靠非特异性吸附作用,导致其吸附容量较小 [24-25]。
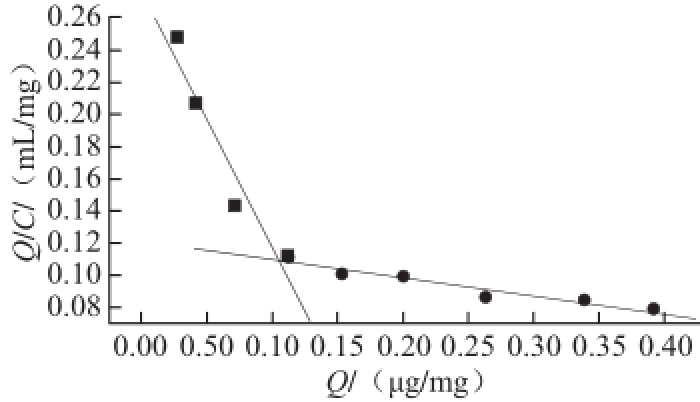
图6 ETP-MIPs的Scatchard曲线
Fig.6 Scatchard plot of ETP-MIPs
如图6所示,通过Origin软件进行拟合,得到两个线性拟合方程,分别为Q/C=0.277-1.608Q(质量浓度范围为0.1~1 μg/mL,R 2=0.958)和Q/C=0.120-0.109Q(质量浓度范围为1~5 μg/mL,R 2=0.965),这表明该分子印迹聚合物中存在两种结合位点。根据线性拟合方程的斜率和截距,可求得ETP-MIPs中高亲和位点与低亲和位点的平衡离解常数K d和最大表观结合量Q max,其中K d1=1.608 μg/mL,Q max1=1.101 μg/mg;K d2=0.109 μg/mL,Q max2=0.172 μg/mg。
以硅胶为载体,ETP为模板分子,NOBE为功能单体,EGDMA为交联剂,采用表面MIT制备ETP-MIPs。采用正交试验方法,从ETP-NOBE物质的量比、ETPEGDMA物质的量比和致孔剂种类3 个因素考察,对ETPMIPs的制备条件进行优化。结果表明:3 个因素影响主次顺序为致孔剂种类>ETP-NOBE物质的量比>ETPEGDMA物质的量比;当致孔剂为乙腈,ETP-NOBEEGDMA的物质的量比为1∶2∶20时,所制备的ETP-MIPs吸附效果最好,印迹效果达到2.804。采用红外光谱、DSC以及场发射扫描电镜测试对其分子结构、热稳定性和表面形貌进行了研究。通过对ETP-MIPs的等温吸附曲线进行测定,并进行Scatchard分析,线性拟合后发现存在两类结合位点,求得其高亲和位点与低亲和位点的平衡离解常数K d和最大表观结合量Q max,分别为K d1=1.608 μg/mL,Q max1=1.101 μg/mg;K d2=0.109 μg/mL,Q max2=0.172 μg/mg。研究表明,以NOBE为功能单体,采用表面分子印迹技术制备的ETP-MIPs对ETP具有一定的选择吸附作用,这为MIT用于动物性食品中ETP残留的分离检测提供了理论参考。
参考文献:
[1] KHORRAMI A R, EDRISI M. Synthesis and evaluation of a molecularly imprinted polymer for solid phase extraction of ethopabate from chicken tissue[J]. Separation Science and Technology, 2010, 45(3): 404-412. DOI:10.1080/01496390903409567.
[2] 杨林, 薄永恒, 高迎春, 等. 高效液相色谱法检测鸭、鹅可食组织中乙氧酰胺苯甲酯残留量[J]. 江苏农业科学, 2015, 43(3): 265-267. DOI:10.15889/j.issn.1002-1302.2015.03.087.
[3] NAGATA T, SAEKI M, NAKAZAWA H, et al. Sensitive determination of ethopabate residues in chicken tissues by liquid chromatography with fluorometric detection[J]. Journal-Association of Official Analytical Chemists, 1985, 68(1): 27-28.
[4] NASR J J, SHALAN S, BELAL F. Determination of ethopabate residues in chicken muscles, liver, and eggs after aqueous SDS extraction by micellar liquid chromatography with fluorescence detection with application to baby food[J]. Food Analytical Methods, 2013, 6(6): 1522-1528. DOI:10.1007/s12161-013-9564-1.
[5] 农业部畜牧兽医局. 动物源食品中乙氧酰胺苯甲酯残留检测方法:高效液相色谱法[J]. 中国兽药杂志, 2002, 36(10): 7-8. DOI:10.3969/ j.issn.1002-1280.2002.10.004.
[6] COX G B, SUGDEN K. Determination of pyrimethamine, ethopabate and sulphaquinoxaline in poultry feeding stuffs by highperformance liquid chromatography using a weak cation-exchange column packing[J]. Analyst, 1977, 102(1210): 29-34. DOI:10.1039/ AN9770200029.
[7] TAN H S, RAMACHANDRAN P, CACINI W. High performance liquid chromatographic assay of amprolium and ethopabate in chicken feed using solid-phase extraction[J]. Journal of Pharmaceutical and Biomedical Analysis, 1996, 15(2): 259-265. DOI:10.1016/0731-7085(96)01829-8.
[8] 岳振峰, 康海宁, 陈小霞, 等. 液相色谱串联质谱法测定鸡肉中20 种抗球虫药物多残留[J]. 分析化学, 2012, 40(8): 1262-1266. DOI:10.3724/SP.J.1096.2012.10752.
[9] 刘国艳, 柴春彦, 申兆菊. 电化学传感器法检测动物性产品中残留乙氧酰胺苯甲酯[C]//中国畜牧兽医学会2006学术年会. 武汉: 中国畜牧兽医学会工作部, 2006: 165-169.
[10] 宋伟, 胡艳云, 韩芳, 等. 超高效液相色谱串联质谱法同时测定鸡肉中的二氯二甲吡啶 酚、磺胺类和喹诺酮类药物残留[J]. 色谱, 2013, 31(12): 1161-1166. DOI:10.3724/SP.J.1123.2013.08022.
[11] TAN J, JIANG Z T, LI R, et al. Molecularly-imprinted monoliths for sample treatment a nd separation[J]. TrAC-Trends in Analytical Chemistry, 2012, 39: 207-217. DOI:10.1016/j.trac.2012.05.009.
[12] SCHIRHAGL R, REN K N, ZARE R N. Surface-imprinted polymers in microfluidic devices[J]. Science China Chemistry, 2012, 55(4): 469-483. DOI: 10.1007/s11426-012-4544-7.
[13] LEJEUNE J P. Design and development of chiral and achiral molecularly imprinted stationary phases[D]. Lake Charles: McNeese State University, 2010.
[14] 柳絮飞. 两种苯磺隆分子印迹聚合物膜的制备及其性能研究[D]. 泰安: 山东农业大学, 2010. DOI:10.7666/d.y1786778.
[15] 刘祝娟. 硅胶表面三聚氰胺分子印迹聚合物的制备及其分析应用[D].哈尔滨: 哈尔滨工业大学, 2010.
[16] 胡廷平, 张宴铭, 郑立辉, 等. 硅胶表面苯并噻吩分子印迹聚合物的分子识别与吸附性能[J]. 燃料化学学报, 2010, 38(6): 722-729. DOI:10.3969/j.issn.0253-2409.2010.06.016.
[17] 郑细鸣, 涂伟萍. 分子印迹聚合物结合与识别能力的影响因素[J]. 材料导报, 2004, 18(10): 57-59. DOI:10.3321/j.issn:1005-023X.2004.10.017.
[18] YANG X, ZHANG Z H, LI J X, et al. Novel molecularly imprinted polymers with carbon nanotube as matrix for selective solid-phase ex traction of emodin from kiwi fruit root[J]. Food Chemistry, 2014, 145: 687-693. DOI: 10.1016/j.foodchem.2013.08.114.
[19] LI X J, ZHANG B L, LI W, et al. Preparation and characterization of bovine serum albumin surface imprinted thermosensitive magnetic polymer microsphere and its application for protein recognition[J]. Biosensors and Bioelectronics, 2014, 51: 261-267. DOI:10.1021/ ac70182 4u.
[20] 韩瑞芳. 用双重识别基团分子印迹聚合物富集天然微量蛋白质的研究[D]. 天津: 南开大学, 2009. DOI:10.7666/d.y1592552.
[21] 杨文明. 表面分子印迹聚合物的制备与性能研究及计算机辅助设计[D]. 镇江: 江苏大学, 2013.
[22] XIE H Q, LUO W, LIU D B, et al. Preparation and adsorption characteristics of cordycep in molecularly imprinted polymers[J]. Analytical Letters, 2014, 47(11): 1888-1899. DOI:10.1080/00032719.2 014.888723#.
[23] 杨卫海, 严守雷, 卫晨, 等. 沉淀聚合法制备三聚氰胺分子印迹聚合物微球[J]. 高分子学报, 2010(10): 1163-1169. DOI:10.3724/ SP.J.1105.2010.09347.
[24] HONG Y S, CHEN L G. Extraction of quercetin from herba lysimachiae by molecularly imprinted-matrix solid phase dispersion[J]. Journal of Chromatography B, 2013, 941: 38-44. DOI:10.1016/ j.jchromb.2013.10.002.
[25] 郑毅. 基于介孔材料HMS的分子印迹材料制备及其吸附性能[D].哈尔滨: 哈尔滨工业大学, 2010.
Preparation and Characterization of Surface Molecularly Imprinted Polymers for Selective Adsorption of Ethopabate
ZHANG Lamei, LI Daomin
*, LI Zhaozhou, LIU Erwei, LI Songbiao, CAO Li, GAO Hongli, WANG Fang, HOU Yuze
(College of Food and Bioengineering, Henan University of Science and Technology, Luoyang 471003, China)
Abstract:The novel functional monomer N,O-bismethacryloyl ethanolamine (NOBE) was synthesized and used to prepare molecularly imprinted polymers (ETP-MIPs) for adsorbing ethopabate by surface molecular imprinting technique using silica gel as the support matrix and ethylene glycol dimethacrylate (EGDMA) as the crosslinking agent. Three polymerization conditions including molar ratio of template to functional monomer, molar ratio of template to crosslinking agent and porogen type were optimized using an orthogonal array design. The type of porogen was the most important factor, followed by the ratio of template to functional monomer and ratio of template to crosslinking agent. The best imprinting efficiency was observed for molecularly imprinted polymers with a molar ratio of ETP to NOBE to EGDMA of 1:2:20 usin g acetonitrile as the porogen. The Scatchard analysis indicated that there were two classes of binding sites in ETP-MIPs; the equilibrium dissociation constant (K d) and apparent maximum binding capacity (Q max) for the high affinity sites and low affinity sites were calculated as follows: K d1=1.608 μg/mL, Q max1=1.101 μg/m g; and K d2=0.109 μg/mL, Q max2=0.172 μg/mg, respectively.
Key words:molecularly imprinted polymers; surface molecular imprinting technology; orthogonal array experiment; ethopabate
DOI:10.7506/spkx1002-6630-201604041
中图分类号:TS207.3
文献标志码:A
文章编号:1002-6630(2016)04-0226-07
引文格式:
张腊梅, 李道敏, 李兆周, 等. 乙氧酰胺苯甲酯表面分子印迹聚合物的制备与吸附特性[J]. 食品科学, 2016, 37(4): 226-232.
DOI:10.7506/spkx1002-6630-201604041. http://www.spkx.net.cn
ZHANG Lamei, LI Daomin, LI Zhaozhou, et al. Preparation and characterization of surface molecularly imprinted polymers for selective adsorption of ethopabate[J]. Food Science, 2016, 37(4): 226-232. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201604041. http://www.spkx.net.cn
收稿日期:2015-06-04
基金项目:河南省教育厅科学技术研究重点项目(14B550015);河南科技大学青年科学基金资助项目(2013QN021)
作者简介:张腊梅(1989—),女,硕士研究生,研究方向为食品安全检测。E-mail:zhanglamei89@foxmail.com
*通信作者:李道敏(1966—),女,副教授,硕士,研究方向为食品质量与安全。E-mail:dmli16@126.com