
图1 不同溶剂儿茶素类标准溶液的高效液相色谱图
Fig.1 Chromatogram of catechins standards in different solvents
郭 颖1,2,黄峻榕1,*,陈 琦2,3,吴雪原2,程 浩2,吴 琼2
(1.陕西科技大学食品与生物工程学院,陕西 西安 710021;2.黄山出入境检验检疫局,安徽 黄山 245000;3.合肥工业大学,安徽 合肥 230000)
摘 要:目前对茶叶中的5 种儿茶素进行分离测定一般采用GB/T 8313—2008《茶叶中茶多酚和儿茶素类含量的检测方法》中的方法,但在实际测定过程中,发现测定结果的重复性较差,空白、标准溶液与样品的溶剂不一致,标准溶液配制过程中标准品不能完全溶解,对国标中儿茶素的测定方法进行改进,用10 mL预热过的体积分数70%甲醇溶液在70 ℃条件下重复提取3 次,合并提取液,用水定容。利用高效液相色谱仪在波长278 nm处进行检测。流动相A为体积分数0.1%乙酸溶液,流动相B为乙腈,洗脱程序为A的初始比例为95%,5 min内A的比例降至90%,保持至15 min,15~20 min内A的比例降至80%,20~25 min内A的比例升至90%,25~30 min内A的比例恢复至95%。结果表明:采用本方法测定茶叶中5 种儿茶素含量可以保证标准溶液与样品溶剂的一致性, 提取较完全,减少了因操作带来的误差,重复性较好,其相对标准偏差为0.75%~4.50%,分离度为0.58~4.35,方法的回收率为81.4%~113.0%,其相对标准偏差均在1.21%~7.71%之间。
关键词:茶叶;儿茶素;测定方法;改进
郭颖, 黄峻榕, 陈琦, 等. 茶叶中儿茶素类测定方法的优化[J]. 食品科学, 2016, 37(6): 137-141. DOI:10.7506/spkx1002-6630-201606024. http://www.spkx.net.cn
GUO Ying, HUANG Junrong, CHEN Qi, et al. Optimization of sample preparation and HPLC chromatographic conditions for the determination of catechins in tea[J]. Food Science, 2016, 37(6): 137-141. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201606024. http://www.spkx.net.cn
绿茶中物质复杂,与茶叶感官品质相关的主要有氨基酸、茶多酚、咖啡碱和糖类,茶多酚是绿茶中产生涩味的主要成分[1-5],同时也是茶叶中主要的活性物质,对人体健康有一定的作用[6-9]。儿茶素类是茶多酚中的主要成分,约占茶多酚总量的70%,其水溶液具有收敛性的涩味,尤其是表没食子儿茶素没食子酸酯(epigallocatechin gallate,EGCG)、表没食子儿茶素(epigallocatechin,EGC)和表儿茶素没食子酸酯(epicatechin gallate,ECG)对茶叶苦涩味的贡献较大[10-12],在茶叶品质的研究中,儿茶素类含量的测定至关重要。目前,茶叶中5种儿茶素类的分离测定主要采用GB/T 8313—2008《茶叶中茶多酚和儿茶素类含量的检测方法》中的高效液相色谱法[13],但是在实际测定过程中存在着一些问题:国标中的标准品的溶剂为水,空白为甲醇,样品的溶剂为体积分数70%甲醇溶液,标准品与空白及样品的溶剂不一致,导致标准品与样品的出峰时间不一致;国标中儿茶素类储备液的浓度超出了儿茶素类在水中的溶解度,且储备液放置一段时间后不溶物会增加;国标中提取方法是用5 mL的体积分数70%甲醇溶液提取2 次,提取不完全,测定结果的重复性较差;EGCG和表儿茶素(epicatechin,EC)的分离效果不是很好。
因此本研究针对以上问题,对流动相A体积分数、溶剂、提取液体积,提取次数和提取条件进行了探讨和实验,对茶叶中儿茶素类含量测定的方法进行了改进。
1.1 材料与试剂
黄山毛峰(2015年4月)采自黄山谢裕大茶叶基地;乙腈、甲醇(均为色谱纯) 美国Tedia公司;乙酸(分析纯) 国药集团化学试剂有限公司;乙二胺四乙酸(ethylenediaminetetraacetic acid,EDTA,分析纯)上海麦克林公司;儿茶素(catechin,C)标准品(纯度≥99%)、EC标准品(纯度≥99%)、ECG标准品(纯度≥99%)、EGC标准品(纯度≥99%)、EGCG标准品(纯度≥99%) 上海源叶生 物科技有限公司。
标准品储备溶液:分别准确称取标准品C、EC各0.2 g,EGC、ECG和EGCG各0.1 g于100 mL容量瓶中,用适量溶剂溶解,用水定容至刻度,放于4 ℃冰箱中备用。
1.2 仪器与设备
LC-20 A高效液相色谱仪(配有紫外检测器及LC solution数据处理系统) 日本岛津公司;Mettler-AL204-IC分析天平(感量0.000 1 g) 美国Mettler公司;MSE225S-000-DU分析天平(感量0.01 mg) 德国Sartorius公司;Vottex-Genie 2可调速旋涡混合器美国SI仪器公司;HH-6数显恒温水浴锅(70 ℃) 上海浦光公司;Hettich Universal 320R离心机(3 000 r/min)德国Hettich公司。
1.3 方法
1.3.1 标准品溶剂的改进
国标中标准品的溶剂为水;本实验中本着溶剂一致性的原则,将溶剂改为体积分数70%甲醇溶液。分别精确称取5 种儿茶素标准品1.00 mg于10 mL容量瓶中,用适量的水和体积分数70%甲醇溶液溶解并用水定容,标明标记,于4 ℃冰箱中保存。
1.3.2 流动相A的改进
国标中的流动相A为体积分数2%乙酸溶液、EDTA溶液:在试剂瓶中加入乙酸20 mL、EDTA溶液2 mL,加水至刻度,摇匀,过膜;流动相中酸的体积分数会对分离效果有一定的影响,改进的方法中流动相A为体积分数0.1%乙酸(加入EDTA体积仍为2 mL)溶液。其他条件保持一致。
1.3.3 样品提取的改进
国标中是在0.2 g样品中加入5 mL在70 ℃水浴锅中预热过的体积分数70%甲醇溶液,提取2次,最后用体积分数70%的甲醇溶液定容到10 mL,提取过程在试管未密封条件下进行;本实验是加入10 mL预热过的体积分数70%甲醇溶液,提取3 次,最后用水定容到50 mL,为了抑制加热过程中儿茶素类的氧化,提取过程是用具塞离心管进行提取,在密封条件下进行。称取0.2 g(精确至0.000 1 g)试样于50 mL离心管中,加入10 mL在70 ℃水浴锅中预热的体积分数70%甲醇溶液提取,放入70 ℃水浴锅中提取10 min(为了使提取更加充分,5 min时振荡一次)。取出后于3 000 r/min离心10 min,吸取上清液于容量瓶中,定容,待用。
1.3.4 样品的净化
取适量的样品提取液用水稀释10 倍,经0.22 μm水系滤膜过滤至进样瓶中,供高效液相色谱分析。
1.3.5 色谱条件
色谱柱:Agilent zorbax SB-C18石英毛细柱(4.6 mm×250 øm,0.5 øm);流动相B:乙腈;流动相A:不同体积分数的乙酸溶液(0.1%、1.0%、2.0%、2.5%,EDTA体积均为2 mL);洗脱程序:A的初始比例为95%,5 min内A的比例降至90%,保持至15 min,15~20 min内A的比例降至80%,20~25 min内A的比例升至90%,25~30 min内A的比例恢复至95%;流速1 mL/min,进样量10 øL。检测器:紫外检测器;检测波长:278 nm。
2.1 标准品溶剂的改进
儿茶素类,易溶于热水、含水乙醚、含水乙醇、醋酸乙酯、丙酮等溶剂中,但不溶于苯、氯仿、石油醚等有机溶剂[14]。国标中儿茶素类标准溶液的配制,使用的是蒸馏水,儿茶素类的溶解性较差,有不溶物存在,且随着存放时间的延长,不溶物含量增加。而用体积分数70%甲醇溶液配制的儿茶素类标准溶液,儿茶素类完全溶解,放置较长时间也不会有沉淀析出。如图1所示,水和体积分数70%甲醇溶液配制的儿茶素类标准溶液中,C、EGC、EC和EGCG的峰高、峰面积基本没变化,但是对于ECG来说,用体积分数70%甲醇溶液配制的溶解性明显优于用水配制的。原因可能是ECG在冷水中的溶解性较在甲醇中的溶解性小。故实验应用体积分数70%甲醇溶液来配制儿茶素类标准溶液,标准曲线的准确性提高,定量分析的结果将会更加准确。
图1 不同溶剂儿茶素类标准溶液的高效液相色谱图
Fig.1 Chromatogram of catechins standards in different solvents
2.2 流动相A的改进
表1 不同体积分数乙酸的流动相A对儿茶素类分离影响的数据比较
Table 1 Effects of different concentrations of acetic acid in mobile A on the separation of catechins
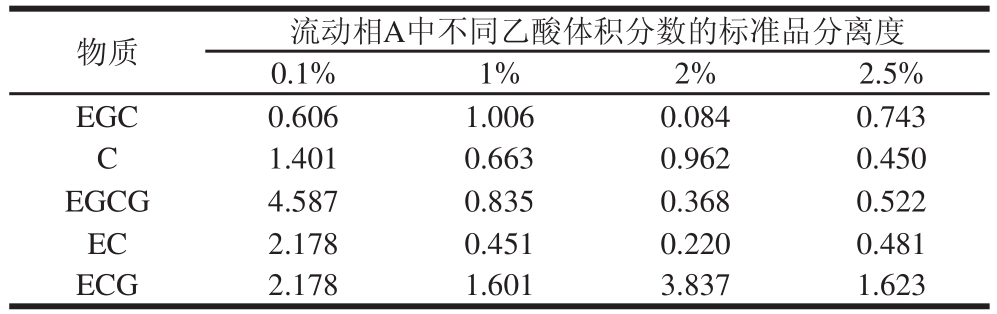
由于儿茶素类结构中有大量的酚羟基,容易电离,在流动相中加入少量的酸可以抑制酚羟基的解离;另一方面,儿茶素类物质高效液相色谱分离的洗脱体系pH值在2.0~3.0之间,可以使峰形变得尖锐、对称,防止峰的拖尾,分离度也可以得到改善[15-19]。通过国标中的方法及肖俊松[20]和凌云[21]等研究对比,发现酸降为体积分数0.1%时,样品中儿茶素类的分离发生明显的变化,不同乙酸体积分数的流动相A测定的样品中儿茶素类的分离度见表1,可以看出不同体积分数的乙酸对儿茶素类的分离有影响,尤其是EGCG和EC的分离度变化最为明显,这与戴军等[17]的研究结果一致。但是,在实际实验中发现,当流动相A中乙酸体积分数为0.1%时,EGCG和EC的出峰顺序发生了变化,分析原因,EGCG和EC的结构不同,导致两者对酸的敏感程度不一致,从结果可以分析得出EGCG对酸更加敏感。
2.3 样品提取条件的改进
在国标中,样品用5 mL的体积分数70%甲醇溶液提取,重复提取1 次,合并提取液,定容至10 mL。由于儿茶素类浓度较高,尤其是EGCG[22],为比较2 种方法的提取效果,分别对按照国标中方法提取2 次后的残渣和改进后方法中提取3 次后的残渣进行提取,在保证稀释倍数一定的情况下,比较提取结果的数据如表2所示,发现国标中提取液中儿茶素类的含量仍然比较高,而采用改进后的方法测定的残渣提取液的儿茶素类含量未检出。通过对比,用10 mL体积分数70%甲醇溶液提取3 次,能保证样品中的儿茶素类提取完全,其提取方法的提取程度明显优于国标中的提取方法。
表2 采用国标中原方法和改进后方法测定儿茶素类的数据比较
Table 2 Comparison of results obtained for the determination of catechins using the Chinese National Standard method and the improved method

注:—.未检出。
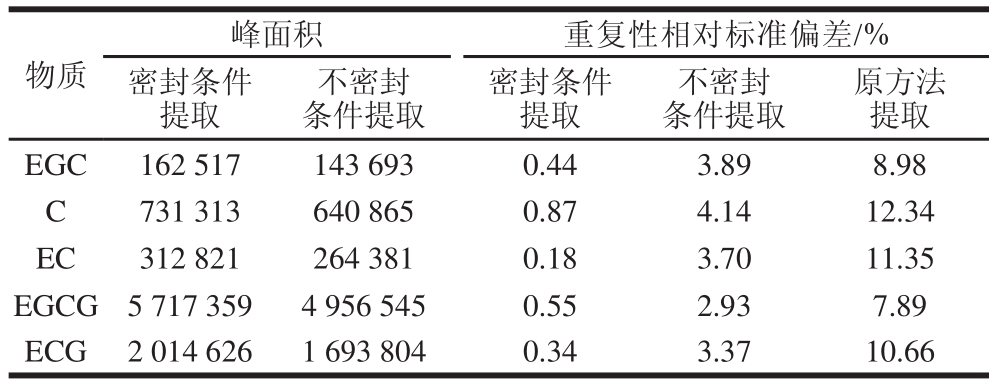
儿茶素类由于自身结构,极易发生氧化、聚合、缩合,酯型儿茶素在湿热条件下,有可能会转化成对映的光学立体异构体,而且水溶液中的酯型儿茶素在一定条件下也可能转化成立体异构体[7]。国标中的用试管进行提取,提取过程与空气直接接触,由于温度高儿茶类素会被氧化[23-24],导致样品测定的重复性较差。实验中用塑料离心管提取,分别在密封和未密封条件下进行,进行5 次平行实验,对结果的重复性进行比较,比较结果见表3。发现密封条件下提取的儿茶素类含量明显高于未密封条件下提取的,说明密封提取有效地减少了空气与茶叶样品提取液的接触,一定程度上抑制了儿茶素类提取过程中的氧化,使提取的结果更加准确。同时对2 种条件下提取的样品提取液进行重复性测定,由表3可以看出,在密封条件下提取的样品的重复性更好。因此,在密封条件下提取儿茶素类的氧化有一定的抑制作用。
表3 密封和不密封条件下提取儿茶素类的数据比较及与原国标中提取稳定性的对比
Table 3 Comparison of sealed and open conditions for the extraction of of catechins with respect to peak area and repeatability (RSD)
2.4 线性范围与检出限
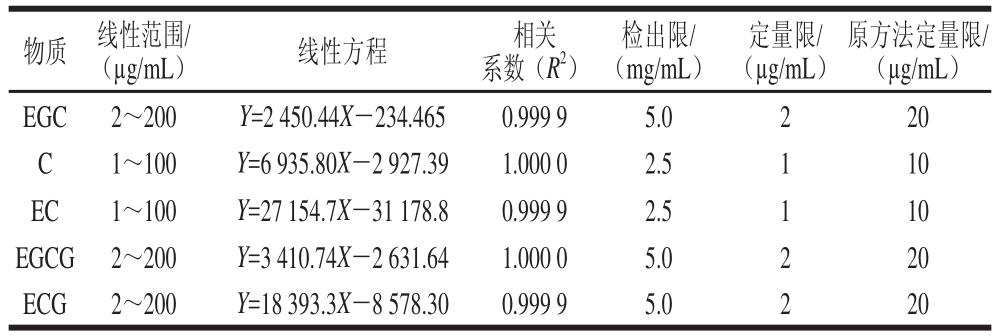
在优化条件下,采用外标法进行定量分析,儿茶素类的标准曲线、线性范围及相关系数见表4。对茶叶样品中5 种儿茶素类含量进行测定,以3 倍信噪比法测得的含量为仪器的检出限,以10 倍信噪比测得的含量为仪器的定量限,结果发现,该方法的定量限明显低于原国标中的方法。
表4 儿茶素类的线性方程、线性范围、相关系数、检出限、定量限
Table 4 Liner equations, liner ranges, correlation coefficients (R2),LODs and LOQs for catechins
2.5 方法的回收率
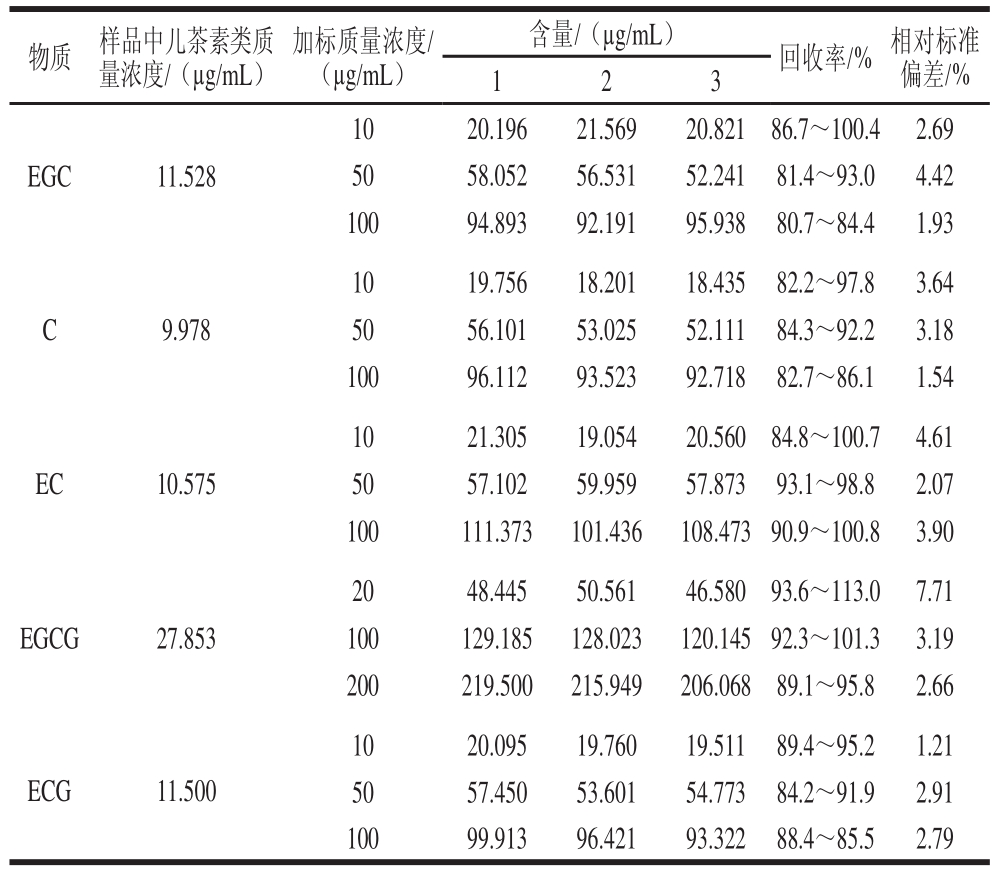
本研究选取一个样品、3 个加标质量浓度进行回收率的测定,每个加标质量浓度进行3 次平行测定,实验结果见表5。该方法的5 种成分、3 个加标质量浓度的回收率均在81.4%~113.0%之间,达到回收率的相关要求,且其相对标准偏差在1.21%~7.71%之间,说明该方法的精确性较好。
表5 儿茶素类含量测定方法的回收率
Table 5 Recoveries of the improved method for catechins determination
2.6 实际样品的测定
选取3 个样品分别进行5 次重复测定,验证方法的稳定性及精密度,按照前述方法进行样品分析,考察方法稳定性的相对标准偏差,结果见表6。在3 个实际样品的测定中,5 种儿茶素类含量的相对标准偏差为0.75%~4.50%,各成分的分离度在0.58~4.35之间,能达到检测的要求。
表6 实际样品儿茶素类含量测定结果的稳定性及分离度
Table 6 Stability and separation degree in the determination of catechins in real samples

本实验对国标中的儿茶素类的测定方法进行了改进,流动相A中酸的体积分数改为0.1%后,儿茶素类的分离效果明显优于原来国标中的,这是由于不同的儿茶素类结构不同,对酸的敏感程度有所不同,这使得儿茶素类的分离程度发生变化。标准溶液用体积分数70%甲醇溶液制备,溶解性较好,稳定性较强。茶叶样品用10 mL的体积分数70%甲醇溶液提取3 次,方法简便、提取程度高、净化效果好、重复性强、有效减少人为误差,能够满足出口茶叶中儿茶素类含量测定的需求。但是,本实验的分析时间为30 min,时间较长,周蓓等[25]对于茶叶中儿茶素类的测定时间仅为15 min,在分析时间这方面后期将进一步研究。另一方面,当乙酸的体积分数变为0.1%时,EGCG和EC的出峰顺序发生了变化,但是两者出峰顺序发生变化的临界乙酸体积分数浓度需要进一步研究。
参考文献:
[1] 施兆鹏, 陈本国, 曾秋霞, 等. 夏季苦涩味的形成与内质成分的关系[J].茶叶科学, 1984, 4(1): 61-62.
[2] 施兆鹏, 刘仲华. 夏季苦涩味化学实质的数学模型探讨[J]. 茶叶科学, 1987, 7(2): 7-12.
[3] 付晓风. 鲜绿茶叶中茶多酚的提取和纯化研究[D]. 南昌: 南昌大学, 2012.
[4] 徐文平, 李大祥, 张正竹, 等. 绿茶几种化学组分苦涩味非线性回归分析及在感官审评中的应用[J]. 茶业科学, 2010, 30(5): 399-406.
[5] 童华荣, 金孝芳, 龚雪莲. 茶多酚感官性质及其对茶叶涩味的影响[J].茶叶科学, 2006, 26(2): 79-86.
[6] EL-SHAHAWI M S, HAMZA A, BAHAFFI S O, et al. Analysis of some selected catechins and caffeine in green tea by high performance liquid chromatography[J]. Food Chemistry, 2012, 134(4): 2268-2275. DOI:10.1016/j.foodchem.2012.03.039.
[7] PERES R G, TONIN F G, TAVARES M F M, et al. Determination of catechins in green tea infusion by reduced flow micellar electrokinetic chromatography[J]. Food Chemistry, 2011, 127(2): 651-655. DOI:10.1016/j.foodchem.2010.12.104.
[8] WEI Kang, WANG Liyuan, ZHOU Jian, et al. Comparison of catechins and purine alkaloids in albino and normal green tea cultivars (Camellia sinensis L.) by HPLC[J]. Food Chemistry, 2012, 130(3): 720-724.
[9] NALDI M, FIORI J, GOTTI R, et al. UHPLC determination of catechins for the quality control of green tea[J]. Journal of Pharmaceutical and Biomedical Analysis, 2014, 88(25): 307-314. DOI:10.1016/j.jpba.2013.08.054.
[10] 金孝芳. 绿茶滋味化合物[D]. 重庆: 西南大学, 2007.
[11] SENANAYAKE S P J N. Green tea extract: chemistry, antioxidant properties and food applications-a review[J]. Journal of Functional Foods, 2013, 5(4): 1529-1541.
[12] KANWAR J, TASKEEN M, MOHAMMAD I, et al. Recent advances on tea polyphenols[J]. Frontiers in Bioscience, 2012, 4(4): 111-131. DOI:10.2741/E363.
[13] 中华全国供销合作总社杭州茶叶研究所. GB/T 8313—2008 茶叶中茶多酚和儿茶素类含量的检测方法[S]. 北京: 中国标准出版社, 2008.
[14] 阮宇成. 茶多酚的组成与茶叶品质[J]. 中国茶叶, 1979(1): 2-5.
[15] 王丽丽, 陈键, 宋振硕, 等. 茶叶中茶多酚检测方法研究进展[J]. 茶叶科学技术, 2013(4): 6-12. DOI:10.3969/ j.issn.1007-4872.2013.04.002.
[16] 刘丽霞. 茶叶中6 种主要儿茶素的高效液相色谱方法建立及应用[D].南京: 南京理工大学, 2013.
[17] 戴军, 王洪新, 陈尚卫, 等. 茶叶及茶多酚中儿茶素的高效液相色谱分析方法研究[J]. 色谱, 2001, 19(5): 398-402.
[18] 郭宇姝, 张沂, 朱新生. 茶多酚的提取分离与含量测定方法进展[J].解放军药学学报, 2007, 23(2): 121-124.
[19] 张国民, 廖予琦, 王庆忠. HPLC内标标准曲线法测定茶叶中4 种儿茶素和咖啡因[J]. 昆明学院学报, 2010, 32(3): 47-50. DOI:10.3969/ j.issn.1674-5639.2010.03.013.
[20] 肖俊松, 袁英髦, 张爱雪, 等. 茶叶中茶多酚和生物碱的测定及聚类和线性判别分析[J]. 食品科学, 2010, 31(22): 343-348.
[21] 凌云, 赵云峰, 李志军, 等. 茶叶及茶饮料中儿茶素和咖啡因的多组分HPLC分析方法[J]. 卫生研究, 2005, 34(2): 187-190.
[22] WU Chunyan, XU Hairong, HÉRITIER J, et al. Determination of catechins and flavonol glycosides in Chinese tea varieties[J]. Food Chemistry, 2012, 132(1): 144-149.
[23] ANANINGSIH V K, SHARMA A, ZHOU Weibiao. Green tea catechins during food processing and storage: a review on stability and detection[J]. Food Research International, 2013, 50: 469-479. DOI:10.1016/j.foodres.2011.03.004.
[24] 钟世安, 周春山, 杨娟玉. 高效液相色谱法分离纯化酯型儿茶素的研究[J]. 化学世界, 2003(5): 237-239; 245; 249.
[25] 周蓓, 王琳, 李伟, 等. 茶叶中甲基儿茶素的分离、纯化和高效液相色谱法分析[J]. 分析化学, 2008, 36(4): 494-498.
GUO Ying1,2, HUANG Junrong1,*, CHEN Qi2,3, WU Xueyuan2, CHENG Hao2, WU Qiong2
(1. School of Food and Biological Engineering, Shaanxi University of Science and Technology, Xi’an 710021, China; 2. Huangshan Entry-Exit Inspection and Quarantine Bureau, Huangshan 245000, China;
3. Hefei University of Technology, Hefei 230000, China)
Abstract: At present, the five catechins in tea are commonly separated and determined according to the Chinese National Standard GB/T 8313—2008, but the results of actual measurement show that the repeatability of this method is poor. Moreover, different solvents are used in blank, standard and sample solutions, and standard substance is not be completely dissolved in the solvent. In this study, an attempt was made to improve these drawbacks. Tea samples are extracted three times at 70 ℃ with 10 mL of 70% preheated methanol solution, and then the extracts were combined and brought up to the final volume with water. The analytes were determined using high performance liquid chromatograph (HPLC) with UV detection at 278 nm. Mobile phase A was 1% acetic acid solution while mobile phase B was acetonitrile. The gradient elution program used was as follows: 0–5 min, 95%–90% A; 5–15 min, 90% A; 15–20 min 90%–80% A; 20–25 min, 80%–90% A; and 25–30 min, 90%–95% A. The results showed that the developed extraction technique that ensured that the solvents of standard solution and samples were consistent allowed complete extraction of catechins, reducing errors arising from the manual handling and having good repeatability. The relative standard deviations (RSDs) for 5 replicate determinations were in the range of 0.75%–4.50%, and the degrees of separation were 0.58–4.35. Recoveries of spiked sample ranged from 81.4% to 113.0%, with RSDs between 1.21% and 7.71%.
Key words: tea; catechin; determination method; improvement
中图分类号:TS207.3
文献标志码:A
文章编号:1002-6630(2016)06-0137-05引文格式:
DOI:10.7506/spkx1002-6630-201606024
*通信作者:黄峻榕(1971—),女,教授,博士,研究方向为淀粉资源的开发与利用、食品添加剂的应用。E-mail:huangjunrong@sust.edu.cn
作者简介:郭颖(1988—),女,硕士研究生,研究方向为茶叶感官品质与成分品质相关性。E-mail:guoying2473@126.com
基金项目:国家质检总局科技计划项目(2014IK119)
收稿日期:2015-06-25