
图1 紫胶树脂钠盐VE微胶囊制备工艺流程图
Fig. 1 Process flow chart for the preparation of vitamin E microcapsules with lac resin sodium
潘正东1,李 凯1,徐 涓1,周梅村2,张 弘1,*,郑 华1
(1.中国林业科学研究院资源昆虫研究所,云南 昆明 650224;2.昆明理工大学化学工程学院,云南 昆明 650224)
摘 要:为探索紫胶树脂钠盐在微胶囊壁材上的应用,将VE作为芯材,采用喷雾干燥法制备VE微胶囊。以包裹率为目标,通过单因素试验和响应面优化试验设计,对制备工艺进行了优化和分析,得到最佳工艺条件为:乳化剂亲水疏水平衡值11、乳化剂用量0.833 g、芯壁比1∶2.2。在此工艺条件下,制得的VE微胶囊为表面光滑的球型颗粒,其包裹率为78.06%,载药量为31.8%。结果表明,以紫胶树脂钠盐为壁材的VE微胶囊具有良好的热稳定性和复溶性,内容物可在中性pH值或肠内环境下释出,是一种较好的新型食品微胶囊壁材。
关键词:紫胶树脂钠盐;VE;微胶囊壁材;喷雾干燥
引文格式:
潘正东, 李凯, 徐涓, 等. 响应面试验优化紫胶树脂钠盐为壁材制备VE微胶囊工艺[J]. 食品科学, 2016, 37(12): 19-26.
PAN Zhengdong, LI Kai, XU Juan, et al. Response surface methodology for optimization of microencapsulation of vitamin E with sodium shellac as wall material[J]. Food Science, 2016, 37(12): 19-26. (in Chinese with English abstract) DOI:10.7506/ spkx1002-6630-201612004. htt p://www.spkx.net.cn
紫胶树脂是寄生在植物上的紫胶虫所分 泌的一种天然树脂,具有热塑性、热固性、抗潮、耐油、耐酸和优良的成膜性能[1-5],广泛应用在军工、电子、医药等行业[6-9],并经美国食品药品监督管理局认证作为食品添加剂[10]。紫胶树脂是由长链羟基脂肪酸和环状倍半萜烯酸组成的聚酯类化合物[11],不溶于水,仅溶解乙醇等少量的有机溶剂,其应用范围受到极大的限制[12-15]。紫胶树脂水溶解性取决于其羧基电离程度,通过碳酸钠与紫胶树脂反应可生成紫胶树脂钠盐,增加了紫胶树脂羧基的电离程度[16]。紫胶树脂钠盐作为新的紫胶树脂基质材料,既保持了紫胶树脂热塑性、成膜性,还兼具有一定的表面活性。
微胶囊技术是利用天然或化学合成的成膜材料将活泼性、敏感性或挥发性的固体或液体物质包裹,得到包裹复合材料,其平均粒径分布在纳米与毫米之间。制备微胶囊的方法有化学法(界面配位法、界面聚合法、原位聚合法以及锐孔-凝固浴法)、物理法(喷雾干燥法、空气悬浮法、超临界流体法、静电结合法、挤压法、包合法、多孔离心法、真空蒸发沉积法、喷雾冷却法、喷雾冷凝法、旋转分离法)以及物理化学法(相分离法、干燥浴法、熔化分散冷凝法、粉末床法)[17-19]等。
疏水性药物VE是一种强有效的自由基清除剂,能保护机体细胞膜生命大分子免遭自由基的攻击,在延缓衰老、防治心血管疾病方面具有良好的效果,但是其暴露在氧、紫外线、碱、铁盐和铅盐条件下会遭受一定程度的破坏[20]。本研究将以紫胶树脂钠盐为壁材、VE为芯材,喷雾干燥方法制备微胶囊,考察紫胶树脂作为微胶囊壁材的可能性。
1.1 材料与试剂
紫胶树脂 中国林业科学研究院资源昆虫研究所特色森林资源工程技术研究中心;碳酸钠、乙醇(均为分析纯) 重庆川东化工有限公司;去离子水 实验室自制;Span-80(分析纯) 天津市风船化学试剂科技有限公司;Tween-80(分析纯) 天津市致远化学试剂有限公司;VE(分析纯) 国药集团化学试剂有限公司。
1.2 仪器与设备
DSX-90数显搅拌机 杭州仪表电机有限公司;Tensor-27傅里叶红外光谱仪 德国布鲁克公司;JC2000A静滴接触角测量仪 上海中晨公司;SD1000喷雾干燥仪 日本东京理化仪器公司;1200高效液相色谱(high performance liquid chromatography,HPLC)仪美国安捷伦科技有限公司;RCZ-8药物溶出仪 天津天光光学仪器有限公司;BZY-1型表面张力仪 上海平轩科学仪器有限公司;T-25高速剪切乳化机 德国IKA公司;FD-1-50冷冻干燥机 北京博医康实验仪器有限公司;BILON92-IID超声波发生器 上海比朗仪器有限公司。
1.3 方法
1.3.1 紫胶树脂钠盐的制备及其表面张力曲线测定
配制浓度为0.1 mol/L碳酸钠溶液,在60 ℃条件下,分别取1 L上述浓度的碳酸钠溶液进行紫胶树脂的溶解,直至溶液饱和,得到紫胶树脂钠盐饱和溶液,进行冷冻干燥后得到固体紫胶树脂钠盐待用[16]。
配制一系列不同浓度的紫胶树脂钠盐样品溶液。在 25 ℃条件下,通过吊片法,在 BZY-1型表面张力仪上测定不同浓度样品溶液的表面张力,重复3 次,取其平均值,得到表面张力与紫胶树脂钠盐浓度之间的关系曲线,其中紫胶树脂钠盐相对分子质量约为500(紫胶树脂是聚合物,其相对分子质量没有确切的值)。
1.3.2 紫胶树脂钠盐为壁材制备VE微胶囊工艺
图1 紫胶树脂钠盐VE微胶囊制备工艺流程图
Fig. 1 Process flow chart for the preparation of vitamin E microcapsules with lac resin sodium
如图1所示,称取一定量的紫胶树脂钠盐,用一定量的蒸馏水将紫胶树脂钠盐溶解在烧杯中,然后按照要求分别称取乳化剂及VE于烧杯中,并与已溶解好的紫胶树脂钠盐溶液混合,得到混合液,用高速剪切乳化机进行乳化7 min后,用超声乳化仪继续乳化,超声乳化结束后,即得微胶囊初乳化液。将制备好的乳状液按照设定的进料流量、进口温度与出口温度进行喷雾干燥,收集产品测定包裹率、载药量。
1.3.3 VE含量测定
HPLC法:色谱柱:C18;流动相:100%甲醇;流量:0.5 mL/min;柱温:30℃;检测波长:284 nm。用VE的标准贮备液配制质量浓度为0.05、0.08、0.1、0.4、0.6、0.8、1.0 g/L的标准溶液,在上述色谱条件下进样5 μL,以质量浓度为横坐标,峰面积为纵坐标做标准曲线。紫外特征吸收见图2。标准曲线方程为:Y=1 892.3X-5.596 1 ,拟合度R2=99.99%。
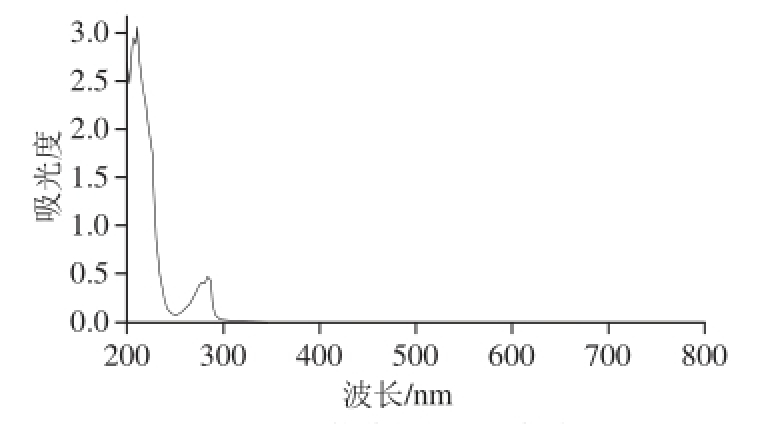
图2 VE的紫外特征吸收峰
Fig. 2 UV characteristic absorption peak of vitamin E
1.3.4 包埋效果的测定[18]
微胶囊表面VE质量测定:取定量微胶囊化产品于烧杯中,添加适量石油醚(沸程30~60 ℃)于烧杯中,进行10 min搅拌,洗下表面油,过滤,收集滤液,在旋转蒸发仪上挥尽石油醚,乙醇定容100 mL,HPLC检测。
微胶囊总VE质量测定:取定量微胶囊化产品于烧杯中,添加适量无水乙醇在37℃的恒温水浴锅中加热溶解,乙醇定容,HPLC检测。
包裹率和载药量计算如式(1)、(2)所示:
1.3.5 单因素试验
亲水疏水平衡(hydrophile-lipophile balance,HLB)值用来衡量乳化剂亲水和亲油性的大小。鉴于实验所用的是Span-80和Tween-80为复配乳化剂,其HLB值选择范围为7~14。配方固定条件:VE用量3 g、乳化剂用量1 g、壁材(紫胶树脂钠盐)用量6 g、芯壁比1∶2(g/g);超声固定条件:超声功率1 000 W、超声时间40 min、超声工作时间1 s(间隔时间2 s);喷雾干燥固定条件:干燥空气(介质)进口温度120 ℃、进料流量0.54 L/h。其他单因素试验在已有单因素试验结果的基础上,保持其他固定条件不变,对制得的VE微胶囊产品进行包裹率、载药量测定。
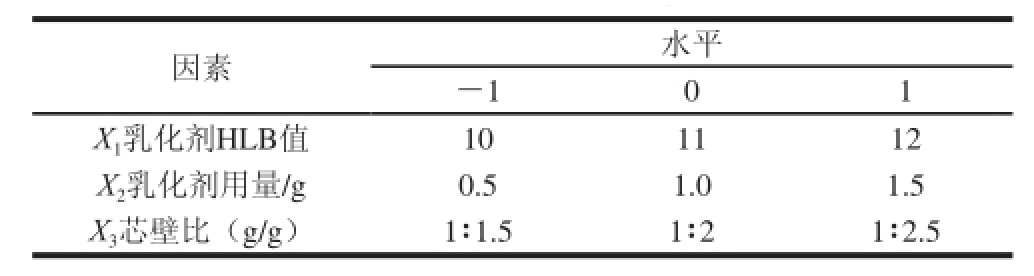
1.3.6 响应面试验设计优化
在单因素试验结果的基础上进行Box-Behnken试验设计,以包裹率为响应值,通过响应面分析对微胶囊条件进行优化。选择对包裹率影响较大的3 个因素乳化剂HLB值、乳化剂用量、芯壁比,借助Minitab 15软件设计程序,对试验进行三因素三水平的设计(表1)。配方固定条件:VE用量3 g、超声功率1 500 W、超声工作时间1 s(间隔时间2 s)、超声时间50 min;喷雾干燥固定条件:干燥空气(介质)进口温度140 ℃、进料流量0.54 L/h。
表1 响应面试验因素与水平
Table 1 levels for the factors used in response surface analysis
1.3.7 VE微胶囊的理化性质检验
1.3.7.1 VE微胶囊的差示扫描量热(differential scanning calorimetry,DSC)分析
VE微胶囊的DSC分析在配置有液氮冷却装置的DSC分析仪上进行。将VE微胶囊样品放入铝盒中,以空铝盒作为参比。对两种样品进行DSC分析,分析条件:初始温度设为0 ℃,以10 ℃/min的升温速率加热至260 ℃,恒温10 min,以保证试样吸热充分和基线平稳。加热过程均在吹扫气和保护气(均为高纯N2,其中吹扫气流速20 mL/min,保护气流速50 mL/min)的气氛下进行,降温介质为液氮。
1.3.7.2 VE微胶囊的扫描电镜表征
由于紫胶树脂钠盐和VE的电阻系数和介电强度高,样品的扫描电镜成像表征首先要进行喷金处理,再在扫描电镜下进行观察、检测及成像。
1.3.7.3 VE微胶囊的红外光谱分析
对紫胶树脂钠盐和VE、VE微胶囊产品分别进行红外光谱检测,其中VE采用液膜法进行检测,其他均采用KBr压片法进行检测。
1.3.7.4 VE微胶囊在不同液体环境中的溶解性质
配制pH 1.0的0.1 mol/L HCl溶液(模拟胃液环境)、pH 6.8磷酸二氢钾-氢氧化钠溶液(模拟肠液环境)、去离子水(pH 7.2)3种不同pH值的溶液1 000 mL,分别称取1 g VE微胶囊(最佳条件下制备,载药量已知)放入药物溶出仪,参照2005年《国家药典》[21]的溶出度检出方法,使用桨法,设定转速为100 r/min,温度为37 ℃,每5 min抽取1 mL溶剂(同时补足新溶剂),测定实时VE质量浓度,从而计算出溶液环境中VE的含量,绘制溶解曲线。
2.1 紫胶树脂钠盐的表面张力
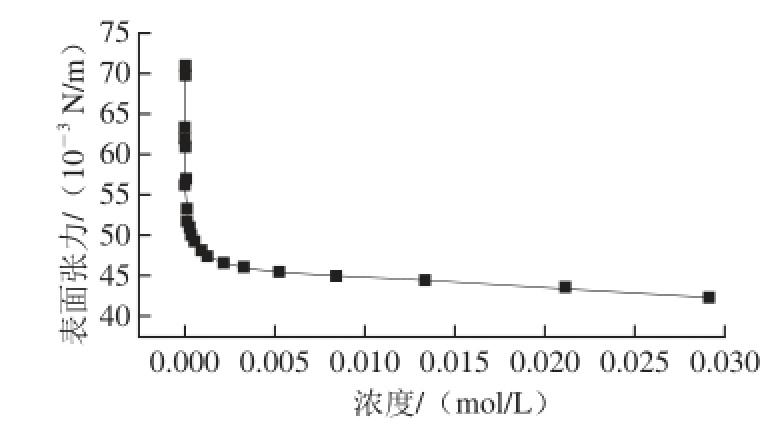
图3 紫胶树脂钠盐表面张力与浓度之间的关系曲线
Fig. 3 Surface tension vs. concentration curve for sodium shellac
如图3所示,紫胶树脂钠盐在一定浓度条件下会发生表面张力显著降低的情况,因紫胶树脂系非均质材料,无普通表面活性剂所具有的明显拐点,但是其降低表面张力的能力还是比较显著。
2.2 单因素试验结果
2.2.1 乳化剂HLB值的选择

图4 乳化剂HLB值与微胶囊包裹率及载药量的关系
Fig. 4 Effect of HLB value of emulsifier on microencapsulation efficiency and drug-loading efficiency
由图4可以看出,随着乳化剂HLB值的增加,包裹率先增加后降低,而载药量在HLB值变化范围内也存在一个最大值,包裹率的变化幅度较大说明乳化剂HLB值是影响包裹率的主要因素。因为乳化剂对喷雾干燥前的初乳液的乳化程度有很重要的影响,乳化程度好,VE被乳化剂和紫胶树脂钠盐包裹后成为细小的微球,微球之间被紫胶树脂钠盐溶液间隔且均匀地分散于乳液中,形成水包油体系;在喷雾干燥过程中以这种微球状态从喷雾口喷出,同时以紫胶树脂钠盐水溶液附着在微球外,在受热后紫胶树脂钠盐溶液固化后成膜包裹含VE的微球,从而达到包裹的目的,试验结果显示,复配乳化剂的HLB值为11时,包裹率和载药量均达到最大值,可以作为下一步单因素试验的固定条件。复配乳化剂的HLB值在10~12范围内VE的包裹率较高,作为下一步响应面法的优化范围。
2.2.2 乳化剂用量的选择

图5 乳化剂用量与微胶囊包裹率及载药量的关系
Fig. 5 Effect of emulsifier dosage on microencapsulation efficiency and drug-loadin efficiency
由图5可以看出,载药量随着乳化剂用量的增加逐渐降低,这是因为随着乳化剂用量的增加,溶液中总的固含量增加,在芯材质量不变的情况下,其比例渐渐降低,造成了VE微胶囊载药量的下降。乳化剂用量在0.5~2.0 g范围时,包裹率急剧先增加后降低;在乳化剂用量较低时不足以使乳化液乳化完全,但是随着乳化剂用量的增加,乳化剂能够自己乳化成球,从而挤占部分壁材,使部分VE微胶囊的包裹性不好,裸露于外,造成了包裹率的降低。试验结果显示,复配乳化剂的用量为1 g时,包裹率达到最大值,可以作为下一步单因素试验的固定条件。乳化剂用量在0.5~1.5 g的范围内包裹率较高,作为下一步响应面法的优化范围。
2.2.3 芯壁比的选择
由图6可以看出,随着壁材用量的增加,载药量急剧降低,降低原因类似于2.2.2节单因素试验,随着芯壁比的减少,溶液中的固含量增加而芯材的质量不变,在整个溶液经过喷雾干燥固化后,使喷出的VE微胶囊载药量逐渐降低;而前期随着芯壁比的减少,VE微胶囊的包裹率逐渐增加,随着壁材量的增加,易形成大块凝胶而不利于VE微胶囊的形成,使部分VE裸露,致其包裹率降低。包裹率在芯壁比为1∶2时达到最大值,可以作为下一步单因素试验的固定条件。芯壁比在1∶1.5~1∶2.5的范围内包裹率较高,可作为下一步响应面法综合优化范围。

图6 芯壁比与包裹率及载药量的关系
Fig. 6 Effect of rate of core material to wall material on microencapsulation efficiency and drug-loading efficiency
2.2.4 超声功率的选择

图7 超声功率与包裹率及载药量的关系
Fig. 7 Effect of ultrasonic power on microencapsueation efficiency and drug-loading efficiency
由图7可以看出,超声功率在500~1 000 W时,载药量有先增加后降低的趋势,但是其仅仅在1%的范围内变化,可见在各个组分固含量的质量比确定后,载药量并没有太大的变化。而包裹率随着超声功率的增加呈线性增加,是因超声功率越大,乳液的乳化效果越好,从而表现出包裹率逐渐增长的趋势。因超声功率超过1 500 W后包裹率随着功率的增长变化不大,所以选取超声功率1 500 W作为单因素试验条件。
2.2.5 超声时间的选择
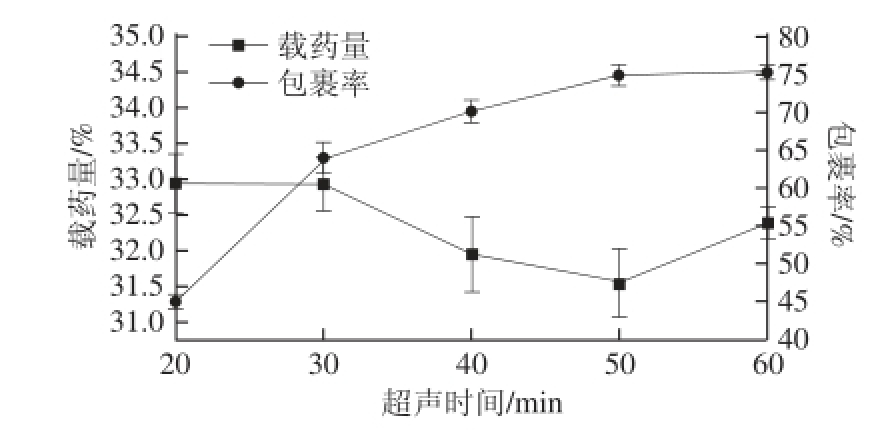
图8 超声时间与包裹率及载药量的关系
Fig. 8 Effect of ultrasonic irradiation time on microencapsulation efficiency and drug-loading efficiency
由图8可以看出,在一定超声时间范围内,包裹率并无太大的变化,随着超声时间的延长,包裹率逐渐增大,但到50 min后,包裹率增加幅度不明显。其对包裹率的作用机理,类似于2.2.4节,故将超声时间50 min作为下一单因素试验的固定条件。
2.2.6 干燥空气(介质)进口温度的选择

图9 干燥空气(介质)进口温度与包裹率及载药量的关系
Fig. 9 Effect of inlet temperature on microencapsulation efficiency and drug-loading efficiency
由图9可以看出,随着干燥空气(介质)进口温度的升高,微胶囊的包裹率也一直在提高,这是因为在高温条件下微胶囊壁能够迅速固化,避免其中的芯材流出,但是当干燥空气(介质)进口温度过高时,水分蒸发速度快,囊壁表面易产生气孔,同时还会导致壁材成膜性降低,产生糊化,从而使包裹率和载药量均下降。由图9可以看出,干燥空气(介质)进口温度在140 ℃为宜。
2.2.7 干燥空气(介质)出口温度的选择

图10 干燥空气(介质)出口温度与包裹率及载药量的关系
Fig. 10 Effect of outlet temperature on microencapsulation efficiency and drug-loading efficiency
干燥空气(介质)出口温度对于微胶囊的囊壁结构和水分含量有较大影响。干燥空气(介质)出口温度高,有利于产品形成完整致密的壁结构,也可使产品水分含量低,有利于改善喷雾干燥的效果。但过高也会导致产品过度受热而开裂,包裹率下降。由图10可以看出,干燥空气(介质)出口温度以66 ℃比较适宜。
2.3 响应面法优化VE微胶囊制备工艺
2.3.1 响应面试验设计与结果
在制备紫胶树脂钠盐VE微胶囊的单因素试验基础上进行Box-Behnken试验设计。以VE微胶囊包裹率作为响应值,选择对其影响较大的3 个因素乳化剂HLB值、乳化剂用量、芯壁比(VE-紫胶树脂钠盐质量比)为变量,并选取这3 个因素中较好水平为考察值,借助Minitab 15软件设计程序对试验进行设计。试验设计与结果详见表2。
表2 Box-Behnken试验设计方案以及微胶囊包裹率测定结果
Table 2 Box-Behnken design with experimental results of microencapsulation efficiency
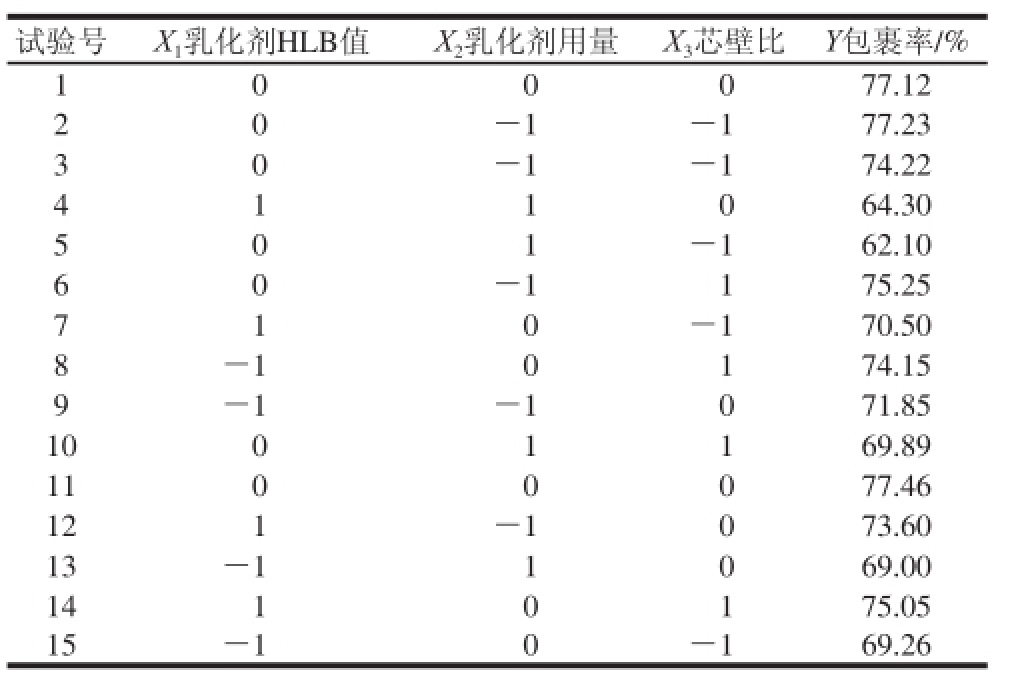
2.3.2 模型方程的建立与显著性试验
根据表2进行试验后,对数据进行二次回归拟合,利用Minitab 15软件建立的数学模型为:

表3 回归方程系数的显著性检验
Table 3 Significance test of regression coefficients in regression model
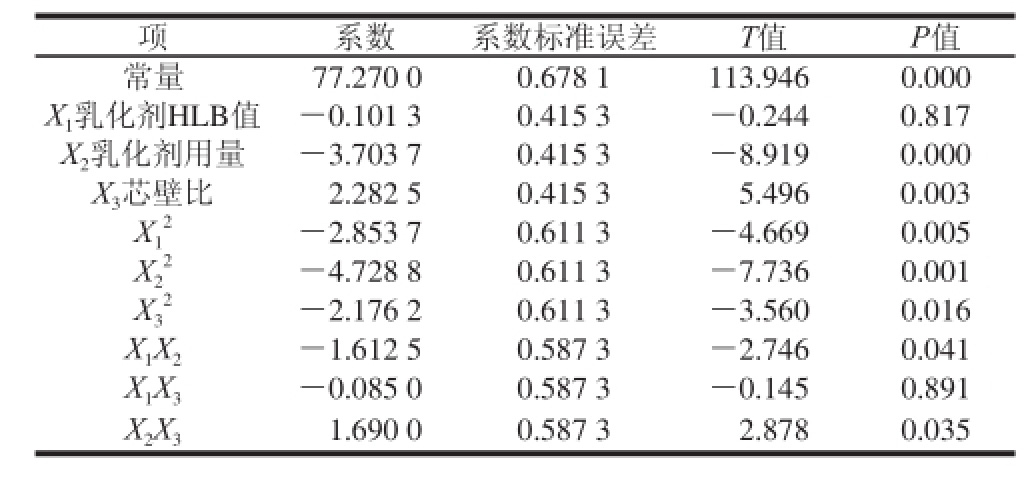
表3表明,根据P值判断,乳化剂用量以及芯壁比依次为影响VE微胶囊包裹率2 个重要的因素,优化试验中乳化剂HLB值重要性并没有明显的体现。
表4 回归模型方差分析
Table 4 Analysis of variance of regression equation
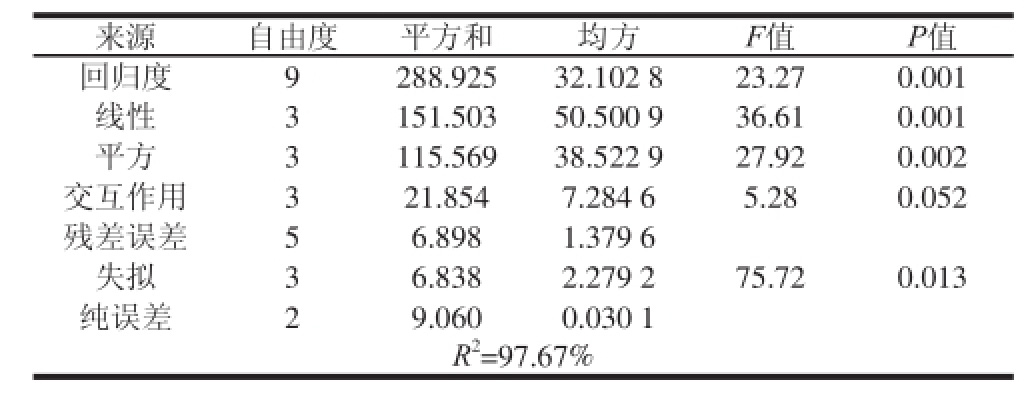
表4方差分析表明,方差线性影响显著,其次是平方项,而交互项影响相对不显著。这是因为乳化剂用量对包裹率影响权重太大,类似于单因素影响,如表4所示,数学模型拟合度为97.67%,拟合情况较好。证明应用响应面法表达VE微胶囊包裹率的模型较为可靠。
2.3.3 响应面优化以及验证实验结果

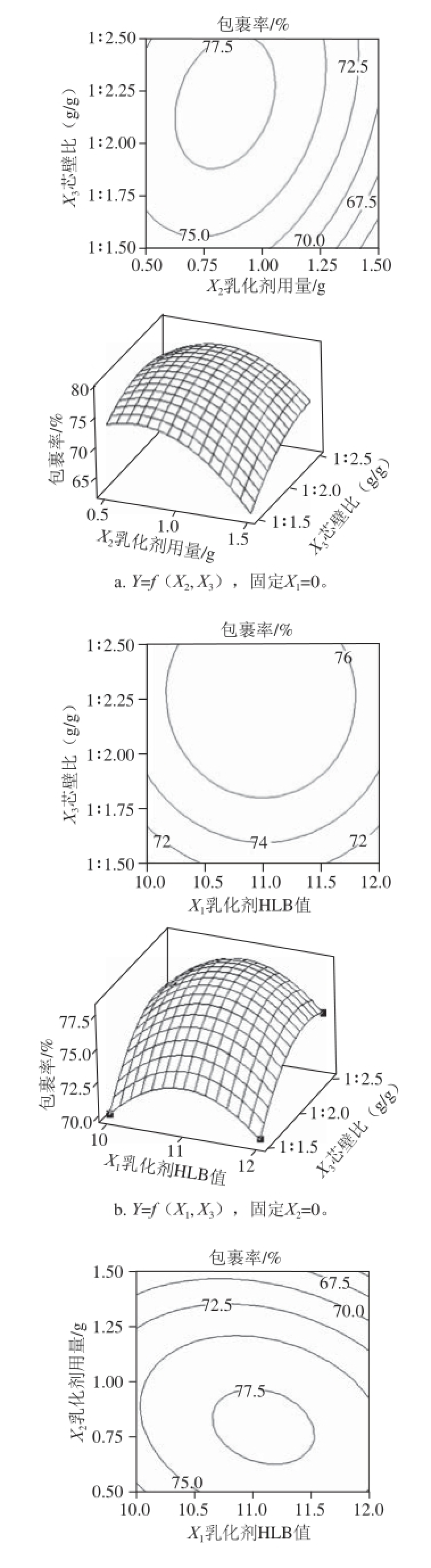

图11 各因素交互作用对包裹率影响的等高线和响应面图
Fig. 11 Response surface and contour plots showing the interactive effects of three factors on microencapsulation efficiency
c. Y=f(X1,X2),固定X3=0。
由图11可以看出,VE微胶囊包裹率较高点均在所选因素范围的中间部分,试验结果较为理想。利用相应Minitab 15软件的响应优化器,以包裹率最大为目标得到优化条件为:乳化剂HLB值11、乳化剂用量0.833 g、芯壁比1∶2.2。进行3 次验证实验,所得微胶囊包裹率分别为77.89%、78.21%、78.08%,其平均值为78.06%,以此优化工艺得到载药量为31.80%。根据数学模型计算的理论预测值为78.33%,与验证实验的平均值78.06%相差不大,说明回归方程能较真实地反映各因素对包裹率的影响。
2.4 VE微胶囊的理化性质检验
2.4.1 VE微胶囊的DSC曲线分析
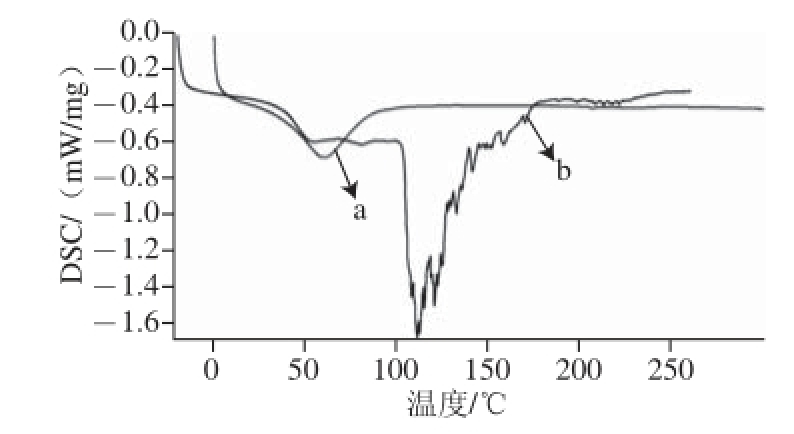
图12 VE微胶囊(a)与紫胶树脂钠盐(b)的DSC分析图
Fig. 12 DSC curves of VE microcapsule and sodium shellac
在微胶囊众多热性质中,玻璃化转变温度是判断微胶囊优异性质的重要的指标,这是因为当贮藏温度高于玻璃化转变温度时,微胶囊的壁材在微观上因分子链开始活动,致使芯材向外扩散和外界物质进入微胶囊的速率增加,从而使产品质量下降[22]。当VE被紫胶树脂钠盐包埋后,VE微胶囊产品的热力学性质可能会发生一定的变化,为了验证微胶囊的形成,采用DSC对产品进行热力学分析。如图12所示,紫胶树脂钠盐制成的VE微胶囊与紫胶树脂钠盐的吸热峰相比明显整体向低温处移动,但是其玻璃化转变温度在48.6 ℃,高于常温,可以在常温条件下贮存使用。
2.4.2 VE微胶囊的扫描电镜分析

图13 以紫胶树脂钠盐为壁材制备的VE微胶囊扫描电镜图
Fig. 13 Scanning electron microphotographs of VE microcapsule
由图13可以看出,本实验制备的微胶囊为球形,表面光滑没有裂缝产生,囊壁的结构保持完整,对芯材保护良好。另外微胶囊的颗粒粒径分布也较为均匀,粒径在3 μm左右。
2.4.3 VE微胶囊红外光谱分析

图14 紫胶树脂钠盐与紫胶树脂VE微胶囊、VE的红外谱图
Fig. 14 Infrared spectra of VE, VE microcapsule and sodium shellac
由图14可以看出,以紫胶树脂钠盐为壁材制成的VE微胶囊红外谱图,保留紫胶树脂钠盐本身的多个特征吸收峰,如:1 566 cm-1处的羧基成盐以后所形成的C=O的振动耦合吸收峰,以及720 cm-1处长链烷烃的特征吸收峰。1 758、1 209 cm-1处属于VE的特征峰在微胶囊的红外谱图中出现,由此可知,紫胶树脂微胶囊是由以上两种物质组成的。
2.4.4 VE微胶囊溶解性分析
向3种不同溶液环境中各加入优化条件制备的VE微胶囊1 g,由于载药量已知为31.80%,1 g VE微胶囊中含有0.318 g VE。在pH 1的模拟胃环境的盐酸溶液中,紫胶树脂钠盐包裹VE微胶囊不溶解,因此也无VE释放。紫胶树脂钠盐在类似于胃液环境的酸条件下不溶解正是其理化特征之一,表明其制备的VE微胶囊具有保药性,使其不在胃中释放内容物,避免其对胃造成刺激。
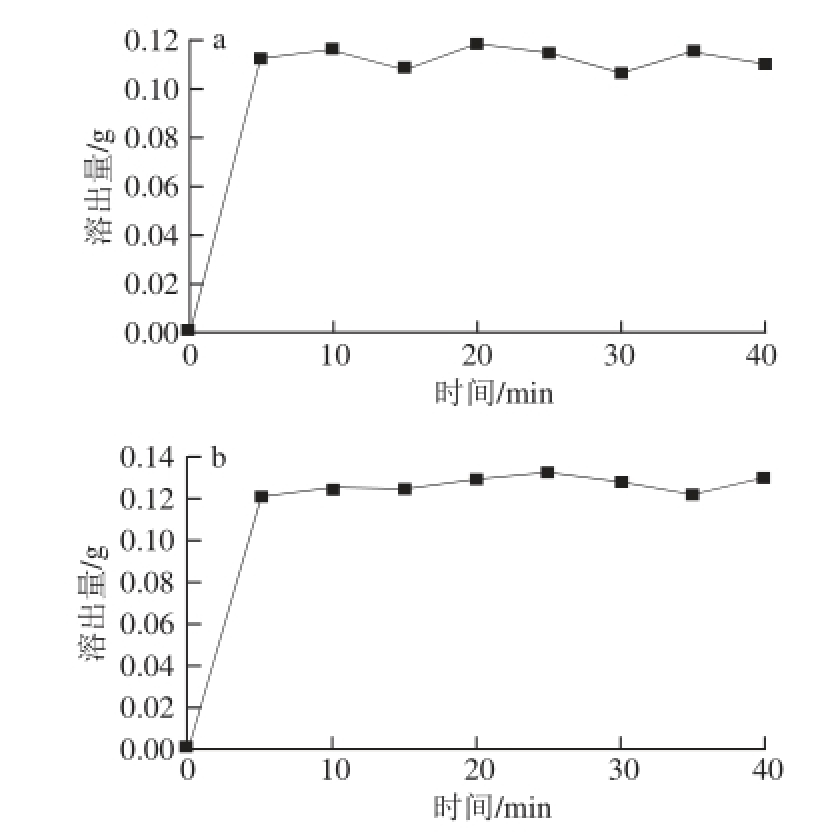
图15 在pH 6.8磷酸盐缓冲液(a)和pH 7.2的去离子水(b)中的VEE释放曲线
Fig. 15 Release curves of VE microcapsule in phosphate buffer solution (pH 6.8) and deionized water (pH 7.2)
如图15a所示,VE微胶囊在溶解5 min后,VE溶出量就达0.112 g(溶出度为35%)。但是在5~40 min,其溶出度并没有大的变化,仅仅是在一定范围内波动。由此可见,紫胶树脂钠盐包裹VE微胶囊溶解性较好,并有突释的特性,但是在实验后期观察到紫胶树脂钠盐VE微胶囊仍有一部分残存、未溶解。去离子水(pH 7.2)中溶解曲线与磷酸盐缓冲液相似,溶解度并没有大的提高,仅在0.12 g附近变化,在实验中观察同样存在未溶解VE微胶囊(溶出度为37%)。
由以上结果可以看出,以紫胶树脂作为壁材的VE微胶囊释放性能一般,其水溶性并不如紫胶树脂钠盐,这是因为在140 ℃条件下经过喷雾干燥过后,一部分壁材受热后,聚合后变为不溶物,并且从图12可以看出,紫胶树脂钠盐在100~150 ℃范围内发生了吸热现象,而在喷雾干燥中经过了140 ℃的进口温度,应有一部分材料受热、聚合变质,致使其溶解性降低。另外,本实验制备的VE微胶囊包裹率有78.06%,尚有21.94%的VE附着在微胶囊的表面,因其为油性物质,一定程度上阻碍了水分与紫胶树脂钠盐的接触,从而降低了VE微胶囊的溶解性。
本研究制备了以紫胶树脂钠盐为壁材的VE微胶囊。以包裹率为目标,通过单因素试验和响应面优化试验设计,对制备工艺进行了优化和分析,得出最佳工艺条件:乳化剂HLB值11、乳化剂用量0.833 g、芯壁比1∶2.2。在此工艺条件下,制得VE微胶囊的包裹率为78.06%,载药量为31.80%。对制得的VE微胶囊进行了理化性质的表征,以紫胶树脂钠盐为壁材的VE微胶囊具有良好的热性质,其玻璃化转变温度为48.6 ℃,高于常温,可以常温贮藏使用;微胶囊为表面光滑的球型微胶囊,粒径分布均匀,在3 μm左右。实验表明紫胶树脂钠盐可以作为VE壁材使用,所得到VE微胶囊成球性较好,复溶性佳,有作为新型食品微胶囊壁材的潜质。
参考文献:
[1] WEI X, PANG J, ZHANG C, et al. Structure and properties of moisture-resistan t konjac glucomannan films coated with shellac/ stearic acid coating[J]. Carbohydrate Polymers, 2015, 118: 119-125. DOI:10.1016/j.carbpol.2014.11.009.
[2] CHITRAVATHI K, CHAUHAN O P, RAJU P S. Postharvest shelflife extension of green chillies (Capsicum annuum L.) using shellacbased edible surface coatings[J]. Postharvest Biology and Technology, 2014, 92: 146-148. DOI:10.1016/j.postharvbio.2014.01.021.
[3] JI H B, ZHANG H, ZHENG H, et al. Improvement of the stability of lac resin and its application[J]. Advance in Forestry Research, 2013, 1: 8-16.
[4] PATEL A R, REMIJN C, CABERO A M, et al. Novel all-natural microcapsules from gelatin and shellac for biorelated applications[J]. Advanced Functional Materials, 2013, 23: 4710-4718. DOI:10.1002/ adfm.201300320.
[5] BELLAN L M, PEARSALL M, CROPEK D M, et al. A 3D interconnected microchannel network formed in gelatin by sacrificial shellac microfibers[J]. Advanced Materials, 2012, 24: 5187-5191. DOI:10.1002/adma.201200810.
[6] 陈奇, 张弘, 郑华, 等. 紫胶树脂在氨水中的溶解性及其铵盐的理化性质[J]. 食品科学, 2013, 34(23): 77-82. DOI:10.7506/spkx1002-6630-201323017.
[7] 甘瑾, 马李一, 陈智勇, 等. 漂白胶对芒果贮藏保鲜效果的研究[J].食品科学, 2005, 26(1): 242-244.
[8] 甘瑾, 张弘, 马李一, 等. 漂白紫胶涂膜对苹果常温贮藏品质的影响[J].食品科学, 2009, 30(23): 444-447.
[9] 甘瑾, 张弘, 马李一, 等. 纳米SiOx漂白紫胶复合膜对椪柑常温贮藏品质的影响[J]. 食品科学, 2009, 30(18): 385-388.
[10] 李凯, 周梅村, 张弘, 等. 漂白紫胶水溶胶的制备[J]. 食品科学, 2009, 30(18): 142-147.
[11] SINGH A, UPADHYE A, MHASKAR V, et al. Chemistry of lac resin-Ⅶ: pure lac resin-3: structure[J]. Tetrahedron, 1974, 30: 3689-3693. DOI:10.1016/S0040-4020(01)90652-0.
[12] 李凯, 张弘, 郑华, 等. 紫胶树脂改性研究进展[J]. 天然产物研究与开发, 2012, 24(2): 274-279.
[13] LUANGTANA-ANAN M, LIMMATVAPIRAT S, NUN THANID J, et al. Effect of salts and plasticizers on stability of shellac film[J]. Journal of Agricultural and Food Chemistry, 2007, 55(3): 687-692. DOI:10.1021/jf061922+.
[14] LIMMAT VAPIRAT S, LIMMATVAPIRAT C, PUTTIPIPATKHACHORN S, et al. Modulation of drug release kinetics of shellac-based matrix tablets by in-situ polymerization through annealing process[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2008, 69(3): 1004-1013. DOI:10.1016/ j.ejpb.2008.01.027.
[15] LIMMAT VAPIRAT S, PANCHAPORNPON D, LIMMATVAPIRAT C, et al. Formation of shellac succinate having improved enteric film properties through dry media reacti on[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2008, 70(1): 335-344. DOI:10.1016/j.ejpb.2008.03.002.
[16] 李凯, 周梅村, 张弘, 等. 紫胶树脂溶解性及其钠盐的理化性质[J].食品科学, 2010, 31(21): 159-164.
[17] 杨小兰, 袁娅, 谭玉荣, 等. 纳米微 胶囊技术在功能食品中的应用研究[J]. 食品科学, 201d 3, 34(21): 359-368. DOI:10.7506/spkx1002-6630-201321076.
[18] 马云标, 周惠明. VE微胶囊的制备及性质研究[J]. 食品科学, 2010, 31(2): 1-5.
[19] 鲍鲁生. 食品工业中应用的微胶囊技术(续)[J]. 食品科学, 1999, 20(11): 6-9.
[20] 杨雷, 仇丹, 周 逸奎, 等. 维生素E微胶囊复水后乳状液的稳定性研究[J]. 食品工业科技, 2014, 35(20): 150-154.
[21] 国家药典委员会. 中国药典[M]. 2部. 北京: 化学工业出版社, 2005: 附录1-5.
[22] 冯岩. 复合凝聚法制备维生素E微胶囊的研究[D]. 无锡: 江南大学, 2008.
Response Surface Methodology for Optimization of Microencapsulation of Vitamin E with Sodium Shellac as Wall Material
PAN Zhengdong1, LI Kai1, XU Juan1, ZHOU Meicun2, ZHANG Hong1,*, ZHENG Hua1
(1. Research Institute of Resources Insects, Chinese Academy of Forestry, Kunming 650224, China; 2. Faculty of Chemical Engineering, Kunming University of Science and Technology, Kunming 650224, China)
Abstract: Sodium shellac, a thermoplastic polymer, has high surfactant activity. In this study, we attempted to fabricate vitamin E microcapsule with sodium shellac as the wall material by spray drying. The optimization of process parameters for improved microencapsulation efficiency was done using one-factor-at-a-time method and response surface methodology. It turned out that the optimal conditions for preparing vitamin E microcapsule were as follow s: hydrophile-lipophile balance (HLB) of emulsifier, 11; emulsifier dosage, 0.833 g; and ratio of core to wall materials, 1:2.2. Under these conditions, the microcapsules with microencapsulation efficiency of 78.06% and drug-loading efficiency of 31.8% were obtained as spherical particles with a smooth surface. The vitamin E microcapsule had high thermal stability and good solubility, and it exhibited good release performance at neutral pH or in the intestinal environment. Thus, sodium shellac potentially could be used in the food industry as a good novel microcapsule wall material.
Key words: sodium shellac; vitamin E; microcapsule wall material; spray drying
收稿日期:2016-02-29
基金项目:国家高技术研究发展计划(863计划)项目(2014AA021801)
作者简介: 潘正东(1990—),男,硕士研究生,研究方向为天然产物化学与利用。E-mail:995645702@qq.com
*通信作者:张弘(1963—),男,研究员,博士,研究方向为林业生物资源化学与利用。E-mail:kmzhhong@163.com
DOI:10.7506/spkx1002-6630-201612004 10.7506/spkx1002-6630-201612004. http://www.spkx.net.cn
中图分类号:TS202.3;TQ351.7
文献标志码:A
文章编号:1002-6630(2016)12-0019-08