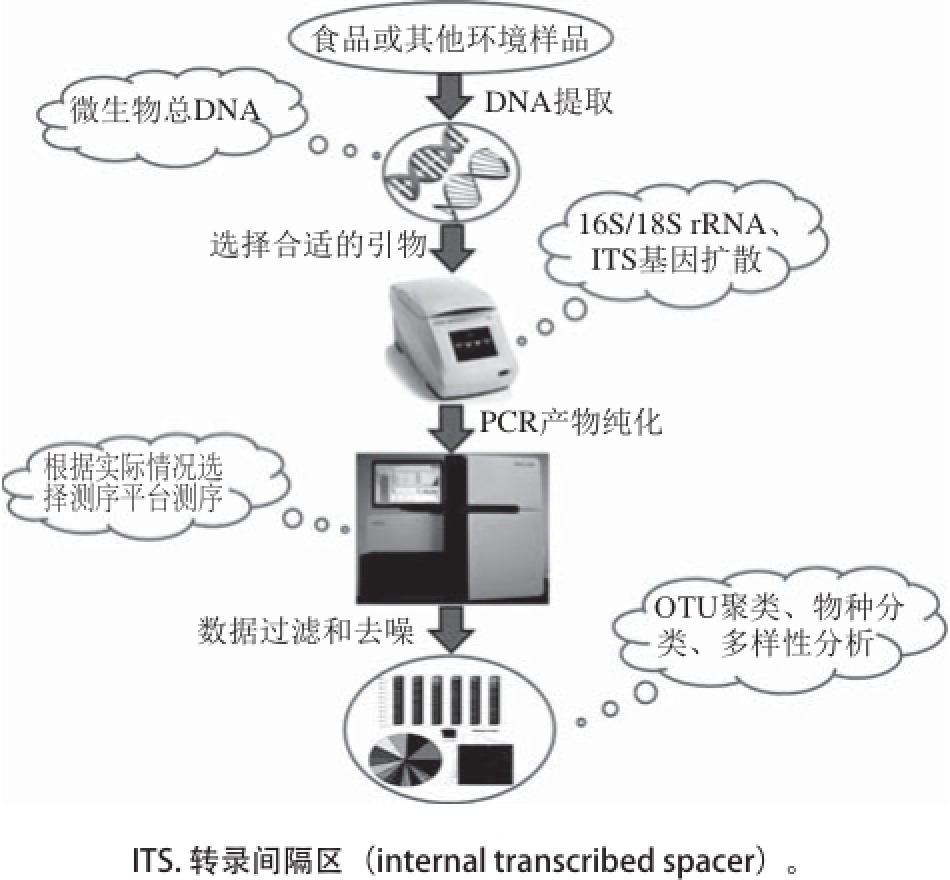
图1 高通量测序应用于食品微生物生态学研究的基本流程
Fig. 1 Overview of the steps involved in high-throughput sequencing in food microbial ecology
米其利 1,2,李雪梅 1,管 莹 1,高 茜 1,桂永发 1,朱洲海 1,夭建华 1,*
(1.云南中烟工业有限责任公司技术中心,云南 昆明 650106;2.云南大学 云南省微生物研究所,云南 昆明 650091)
摘 要:随着分子生物学的发展,越来越多新的研究投入到对食品微生物的研究中。高通量测序作为一种新兴的分子生物学技术,具有数据产出通量高、分析全面、灵敏、快速等特点,被广泛用于食品微生物生态学的研究中。本文对高通量测序的操作流程及其在食品微生物生态学研究中的应用进行总结,评价该技术的优势和局限性,并对其在食品微生物研究中的应用前景进行展望,以期为食品微生物生态学的研究提供参考。
关键词:食品微生物生态学;高通量测序;多样性;微生物区系
微生物是食品生产的基础,同时也是食品污染和腐败产生的关键因素,关系着食品的质量和安全,受到了食品研究者的广泛关注。长期以来,食品微生物生态学的研究都以微生物的纯培养为基础,且研究者们陆续从各种食品中分离鉴定了多种微生物 [1-3]。但是由于培养基和培养条件的限制,只有不到1%的微生物能够通过常规纯培养方法分离出来,仅占环境中微生物的极小部分,很多自然发酵食品中的微生物群落结构仍然无法认识 [4]。为了进一步了解食品微生物生态,保障食品质量和安全,各种新兴的分子生物学方法,包括克隆文库构建、变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)、末端限制性片段长度多样性、实时荧光定量聚合酶链式反应(real-time quantitative polymerase chain reaction,RT-qPCR)、宏基因组学以及新一代的高通量测序技术被广泛应用于食品微生物多样性、群落结构变化以及功能微生物的研究中,极大地改变了人们对食品微生物生态学的认识 [4-9]。
高通量测序是最近10 年新兴发展起来的免培养分子生物学技术,又称新一代测序技术,主要包括Roche 454 Life Sciences焦磷酸测序、Illumina Solexa聚合酶测序和ABI SOLiD连接酶测序3 种测序技术 [10-11]。3 种测序技术的原理各不相同,数据产出、数据质量和单次运行成本也不一样。许多研究也对3 种测序技术的优势和不足进行了比较 [12-14]。目前,高通量测序技术已被广泛应用于土壤、根际、植物内部、水体、动物肠道以及食品等多种环境微生物生态的研究中,极大地改变了人们对环境微生物生态学的认识 [8,15-19]。在食品微生物生态学的研究中,高通量测序技术有助于全面了解食品中微生物群落动态和生理活性,以便提高食品质量、控制其微生物安全。本文就高通量测序技术的操作流程、在食品微生物生态学中的应用和局限性进行综述,以期为食品微生物生态学的研究提供参考。
高通量测序技术分析食品微生物群落的主要操作流程包括:1)样品中微生物总DNA或RNA提取;2)引物选择和PCR扩增;3)测序;4)生物信息学分析,包括操作分类单元(operational taxonomic unit,OTU)聚类、物种分类和多样性分析(图1)。
图1 高通量测序应用于食品微生物生态学研究的基本流程
Fig. 1 Overview of the steps involved in high-throughput sequencing in food microbial ecology
1.1 样品中微生物DNA的提取
采用高通量测序技术研究环境中微生物群落结构时,首先要尽可能获得环境中所有微生物的DNA。只有选择合适的DNA提取方法、提高DNA的提取率、尽可能获得整个样品中所有的微生物基因组,才能真实地反映样品中微生物的实际群落组成。提取不同环境样品微生物基因组的方法有很多,包括多种商业化的DNA提取试剂盒,但主要步骤基本相同,包括样品的机械破碎、细胞溶解、固体杂质的去除、核酸的选择沉淀和离心分离、核酸洗涤和溶解。为了降低DNA提取过程对不同样品之间的差异,同一批样品应采用相同的提取方法提取总DNA,尽管样品之间的理化性质和提取效率可能并不相同。对于一些腐殖质含量较高的环境样品,如森林土壤和堆肥,腐殖质的存在将严重影响PCR的扩增,可采用二氧化硅基质材料固定DNA,去除腐殖质,或采用稀释法,即将DNA样品稀释后再进行PCR扩增 [20]。目前,常用于食品微生物DNA提取的方法主要有试剂盒法、十六烷基三甲基溴化铵法以及苯酚/氯仿抽提法 [21]。
1.2 引物选择和PCR扩增
表1 高通量测序技术在食品微生物研究中的应用
Table 1 Applications of high-throughput sequencing in food microbiology
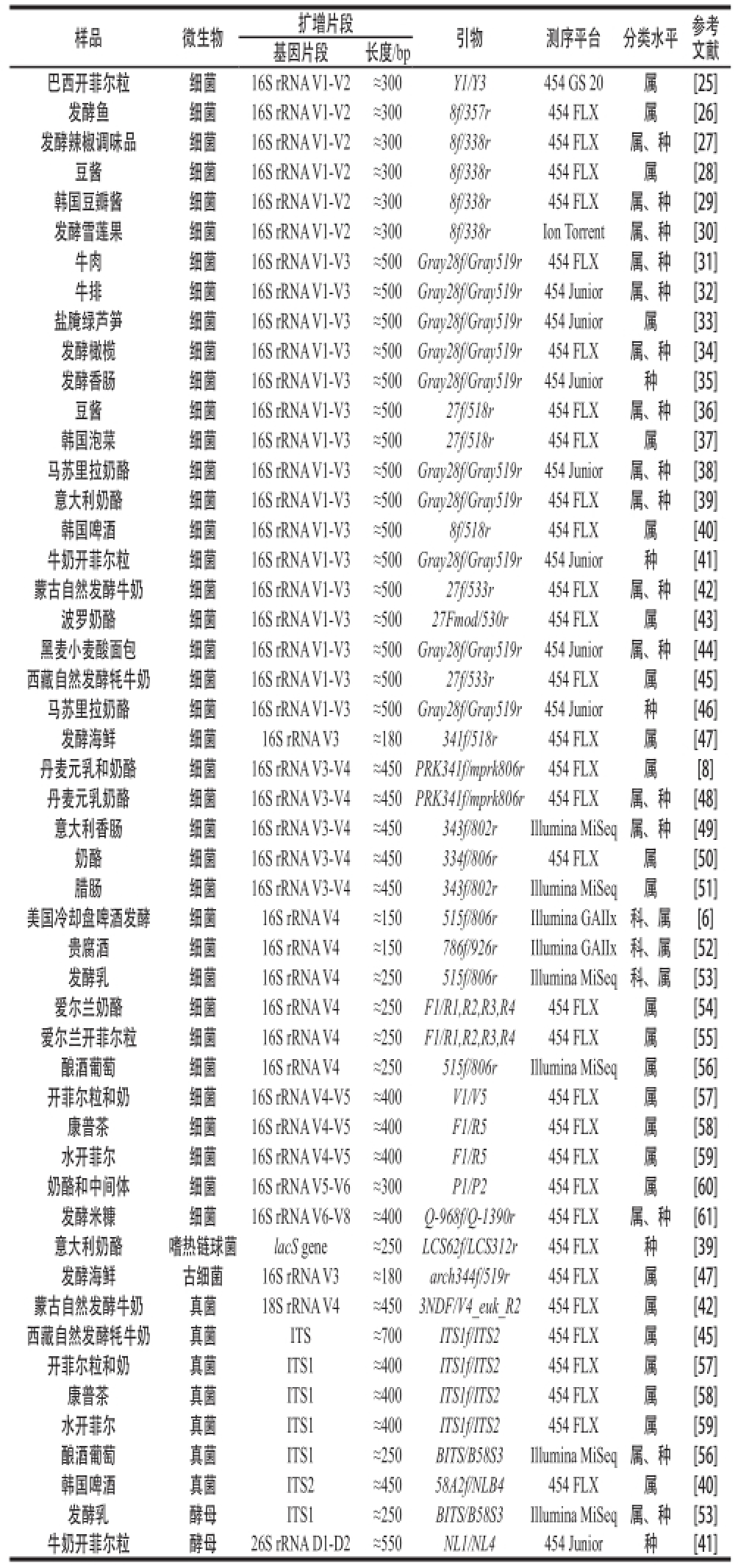
基因片段长度/bp巴西开菲尔粒细菌16S rRNA V1-V2≈300Y1/Y3454 GS 20属[25]发酵鱼 细菌16S rRNA V1-V2≈3008f/357r454 FLX属[26]发酵辣椒调味品细菌16S rRNA V1-V2≈3008f/338r454 FLX属、种[27]豆酱细菌16S rRNA V1-V2≈3008f/338r454 FLX属[28]韩国豆瓣酱细菌16S rRNA V1-V2≈3008f/338r454 FLX属、种[29]发酵雪莲果细菌16S rRNA V1-V2≈3008f/338rIon Torrent属、种[30]牛肉细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 FLX属、种[31]牛排细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 Junior属、种[32]盐腌绿芦笋细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 Junior属[33]发酵橄榄细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 FLX属、种[34]发酵香肠细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 Junior种[35]豆酱细菌16S rRNA V1-V3≈50027f/518r454 FLX属、种[36]韩国泡菜细菌16S rRNA V1-V3≈50027f/518r454 FLX属[37]马苏里拉奶酪细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 Junior属、种[38]意大利奶酪细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 FLX属、种[39]韩国啤酒细菌16S rRNA V1-V3≈5008f/518r454 FLX属[40]牛奶开菲尔粒细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 Junior种[41]蒙古自然发酵牛奶细菌16S rRNA V1-V3≈50027f/533r454 FLX属、种[42]波罗奶酪细菌16S rRNA V1-V3≈50027Fmod/530r454 FLX属[43]黑麦小麦酸面包细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 Junior属、种[44]西藏自然发酵牦牛奶细菌16S rRNA V1-V3≈50027f/533r454 FLX属[45]马苏里拉奶酪细菌16S rRNA V1-V3≈500Gray28f/Gray519r454 Junior种[46]发酵海鲜细菌16S rRNA V3≈180341f/518r454 FLX属[47]丹麦元乳和奶酪细菌16S rRNA V3-V4≈450 PRK341f/mprk806r454 FLX属[8]丹麦元乳奶酪细菌16S rRNA V3-V4≈450 PRK341f/mprk806r454 FLX属、种[48]意大利香肠细菌16S rRNA V3-V4≈450343f/802rIllumina MiSeq属、种[49]奶酪细菌16S rRNA V3-V4≈450334f/806r454 FLX属[50]腊肠细菌16S rRNA V3-V4≈450343f/802rIllumina MiSeq属[51]美国冷却盘啤酒发酵细菌16S rRNA V4≈150515f/806rIllumina GAIIx科、属[6]贵腐酒细菌16S rRNA V4≈150786f/926rIllumina GAIIx科、属[52]发酵乳细菌16S rRNA V4≈250515f/806rIllumina MiSeq科、属[53]爱尔兰奶酪细菌16S rRNA V4≈250F1/R1,R2,R3,R4454 FLX属[54]爱尔兰开菲尔粒细菌16S rRNA V4≈250F1/R1,R2,R3,R4454 FLX属[55]酿酒葡萄细菌16S rRNA V4≈250515f/806rIllumina MiSeq属[56]开菲尔粒和奶细菌16S rRNA V4-V5≈400V1/V5454 FLX属[57]康普茶细菌16S rRNA V4-V5≈400F1/R5454 FLX属[58]水开菲尔细菌16S rRNA V4-V5≈400F1/R5454 FLX属[59]奶酪和中间体细菌16S rRNA V5-V6≈300P1/P2454 FLX属[60]发酵米糠细菌16S rRNA V6-V8≈400Q-968f/Q-1390r454 FLX属、种[61]意大利奶酪嗜热链球菌lacS gene≈250LCS62f/LCS312r454 FLX种[39]发酵海鲜古细菌16S rRNA V3≈180arch344f/519r454 FLX属[47]蒙古自然发酵牛奶真菌18S rRNA V4≈4503NDF/V4_euk_R2454 FLX属[42]西藏自然发酵牦牛奶真菌ITS≈700ITS1f/ITS2454 FLX属[45]开菲尔粒和奶真菌ITS1≈400ITS1f/ITS2454 FLX属[57]康普茶真菌ITS1≈400ITS1f/ITS2454 FLX属[58]水开菲尔真菌ITS1≈400ITS1f/ITS2454 FLX属[59]酿酒葡萄真菌ITS1≈250BITS/B58S3Illumina MiSeq属、种[56]韩国啤酒真菌ITS2≈45058A2f/NLB4454 FLX属[40]发酵乳酵母ITS1≈250BITS/B58S3Illumina MiSeq属、种[53]牛奶开菲尔粒酵母26S rRNA D1-D2≈550NL1/NL4454 Junior种[41]样品微生物扩增片段引物测序平台分类水平参考文献
利用高通量测序技术解析环境微生物多样性,PCR技术属于关键技术,然而PCR结果的可靠性和准确性又受到许多因素的影响,其中以引物选择最为关键。理想的引物应扩增出样品中所有微生物,而且扩增片段长度适宜利于测序且容易区分不同物种之间的差异 [10]。但是已知的引物标记均不能满足上述要求。目前采用高通量测序研究环境微生物生态学所应用的引物多为基于细菌16S rRNA、真菌ITS等编码基因片段设计的特异性引物,但其中难免存在一些问题 [10]。例如,Sun Donglei等 [22]研究了不同引物16S rRNA靶序列扩增对环境微生物多样性影响,指出当采用引物扩增16S rRNA全基因时,将造成环境微生物多样性的过高估计,而采用16S rRNA基因V4-V5区标记时结果最接近真实情况。表1列举了目前应用于食品微生物生态学高通量测序研究中的PCR扩增引物。
在PCR扩增过程中要同时保证扩增效率和扩增产物的特异性,所以需要选择适当的引物和扩增程序保证PCR扩增产物的质量,避免异源双链和单链DNA的形成。同时,在保证扩增产物量的同时应尽量减少PCR循环数,避免嵌合体序列的形成 [23]。对于一些较特殊的环境样品DNA,可采用降落PCR扩增程序,一般可获得较好的扩增结果。在测序前,需要对PCR产物进行纯化和定量,确保各样品DNA浓度一致,保证各样品之间测序结果的均匀分布 [10]。
1.3 测序平台的选择
纯化后的PCR产物经过一系列的前处理后即可进行高通量测序。测序过程因测序平台不同而存在差异。目前,Roche 454 Life Sciences焦磷酸测序平台、Illumina Solexa聚合酶测序平台和ABI SOLiD连接酶测序平台3 种高通量测序平台广泛应用于微生物生态学的研究中。由于这些测序平台测序原理不同,所以在测序深度、读长、运行时间、错误类型以及测序成本等方面均有一定的差异,可以根据自己的研究需要进行选择 [10-11]。许多文献都对这些测序平台的原理和性能比较做了大量的报道,比如Roche 454 Life Sciences焦磷酸测序序列读长在3 种测序平台中最长(600~1 000 bp),但其测序通量最低(0.5~1.0 GB)、测序费用较高 [11]。目前在绝大部分环境微生物生态学相关的研究中多采用这种测序平台。Illumina Solexa聚合酶测序通量大,该测序平台目前主要有HiSeq技术和MiSeq技术,其中HiSeq 2000在1 个运行周期后可产出600 GB的数据量,而且错误率很低,仅为0.26%,MiSeq技术错误率为0.8% [24]。HiSeq技术和MiSeq技术两者检测出的序列读长可分别达到2×150 bp和2×250 bp,可基本满足需要 [24]。ABI SOLiD连接酶测序平台读长最短(50 bp),但其创新之处在于双碱基编码的应用,极大地降低了测序的错误率,由于其双碱基编码和校正系统原理与重测序相似,因此该测序平台多应用于具有高质量参考基因序列物种的重测序,而很少应用于微生物生态学的研究 [10]。在食品微生物生态学研究中,Roche 454 Life Sciences焦磷酸测序(特别是454 FLX技术)和Illumina Solexa聚合酶测序(特别是MiSeq技术)得到广泛应用(表1)。
1.4 生物信息学分析
高通量测序获得的海量数据需要通过生物信息学分析翻译成具体的群落多样性信息。生物信息学分析是高通量测序研究环境微生物生态学研究的重点和难点,涉及到一系列工具和资源,如数据库、分析软件、门户网站和一些网络服务。内容主要包括数据预处理、测序错误校正、OTU聚类、物种分类、α多样性分析和β多样性分析。具体操作可参考Lindahl等 [10]的总结和一些商业测序公司提供的数据处理方法。
高通量测序应用于食品微生物生态学的研究主要集中在食品发酵和腐败微生物区系结构和时空动态变化特征,包括细菌、真菌、酵母以及古菌(表1)。食品发酵过程中微生物群落结构的动态规律直接影响着发酵食品产品的风味和质量,一些病原微生物的繁殖还可能带来潜在的食品安全隐患。因此,监测食品发酵过程中微生物群落结构组成变化特征对于发酵食品安全生产具有重要意义。高通量测序能够同时分析多个样品,被广泛应用在动态监测食品发酵过程中微生物多样性和群落结构组成的研究中。Greppi等 [35]研究了不同发酵阶段的意大利腊肠中细菌群落结构变化,结果表明,在发酵初始阶段(0 d),热杀索丝菌、发光杆菌和莓实假单胞菌为优势类群,随着发酵时间的增长,这3 类细菌含量均急剧降低,发酵时间3~35 d内均保持在较低水平。相反,发酵时间3~35 d内,弯曲乳杆菌、清酒乳杆菌及肉色明串珠菌含量急剧升高,直到发酵结束均为优势菌群,特别是清酒乳杆菌,在整个发酵阶段含量均高于50%,为绝对优势类群。随着发酵时间的延长,腊肠中微生物的多样性指数逐渐增大,微生物多样性更高。基于16S rRNA基因V1-V3区焦磷酸测序,Ercolini等 [44]采用Cytoscape平台(http://cytoscape.github.io/)构建了研究了黑麦、硬质小麦以及普通小麦3 种面粉发酵过程中细菌群落演化的网络关系。结果表明,3 种类型的面粉均包含非常复杂的微生物群落组成,且相互之间微生物类群差异巨大,共有的细菌类群仅占很小部分。但是,随着发酵的开始,微生物多样性急剧降低,相互之间共有的细菌类群所占比例增加,到发酵后期(10 d),3 种来源的发酵面团中微生物组成已十分相似(图2)。
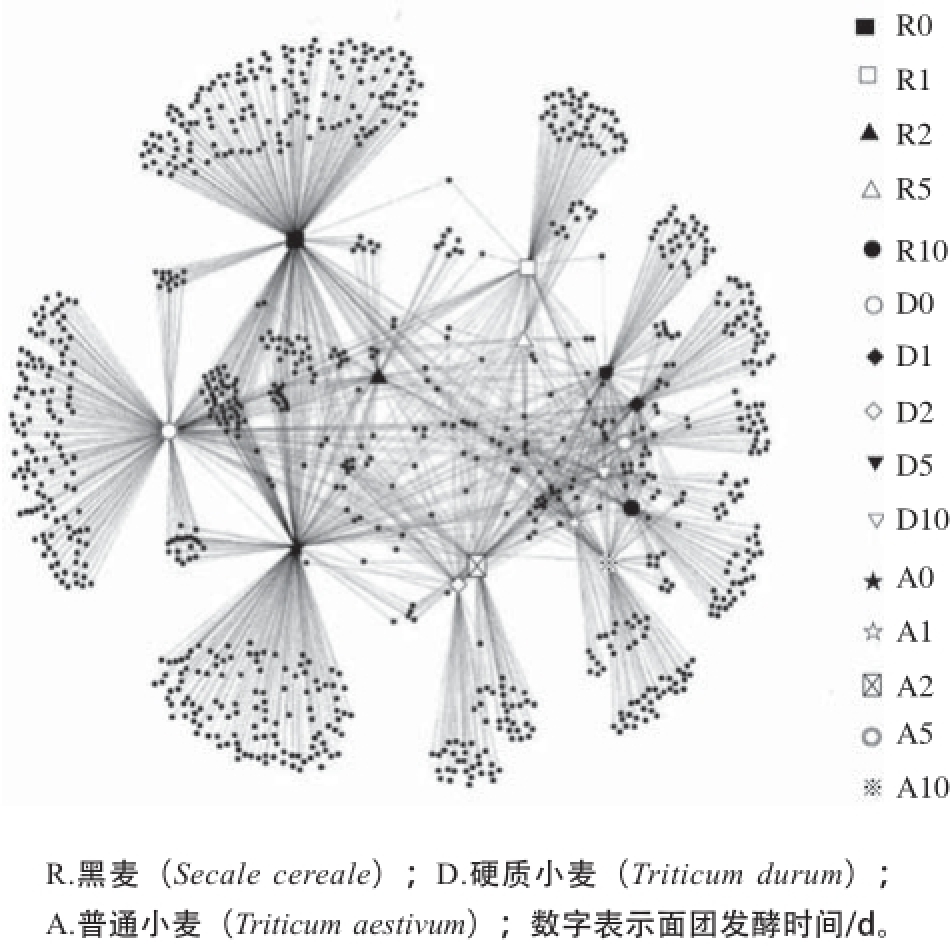
图2 基于16S rRNA基因焦磷酸测序构建的发酵面团微生物网络图
[[4444]]
Fig. 2 Network diagrams of the microbial evolution of three sourdoughs based on 16S rRNA gene data obtained by pyrosequencing
[44]
食品生产原料和生产工艺对微生物群落结构存在显著的影响。采用Illumina高通量测序研究猪肉、牛肉和羊肉腊肠中细菌的多样性,结果显示不同动物来源的腊肠细菌多样性存在显著差异 [51]。Quigley等 [54]采用高通量测序调查了62 份不同原料、不同熟化程度以及不同制作流程的爱尔兰手工干酪和干酪皮中细菌群落的多样性,结果表明干酪微生物区系结构随奶源和配料种类不同而不同,与牛奶是否通过巴斯德消毒有关。同时,研究发现乳酸菌(lactic acid bacteria,LAB)种群结构与干酪熟化程度水平存在显著相关性,硬质干酪LAB种群数量显著高于软质干酪。一些微生物类群,如栖粪杆菌属、普雷沃氏菌属和创伤球菌属首次在干酪中发现,节杆菌属和小短杆菌属也首次发现在山羊奶制作的干酪中。此外,研究还发现明串珠菌属和假单胞菌属2 类细菌仅存在盐浓度较低的样品中,而在高盐含量的干酪样品中并不存在。大量研究结果表明,微生物群落结构的复杂程度与食品生产方式存在密切的关系,越是传统的制作工艺,生产的食品中微生物的群落结构就越复杂,相反,工业化生产的食品一般微生物群落结构相对单一 [27-29]。
食品中各个部分的微生物群落结构也存在一定的差异,且对食品的成熟和腐败具有重要影响。Ercolini等 [62]采用PCR-DGGE研究了斯蒂尔顿干酪不同部位的微生物群落结构,指出干酪表皮和内部微生物群落结构存在显著差异,如植物乳杆菌仅存在于干酪表皮下,而明串珠菌属微生物则均匀分布在干酪的各个部位。为了深入评估食品不同部位的微生物区系差异,高通量测序也被应用于这类研究中。例如,Quigley等 [54]采用高通量测序比较了干酪内部和表皮细菌群落结构差异,结果发现一些以前从未在干酪皮上发现过的微生物类群,如普雷沃氏菌属、法克兰氏菌属以及弧菌属在干酪皮中大量存在,极大地加深了人们对干酪皮微生物区系的认识,也体现出高通量测序技术在认识食品微生物区系的优越性。
高通量测序也被用于监测食品贮藏过程中微生物的变化特征。监测食品贮藏过程中微生物的群落变化特征对于建立适当的食品贮藏条件、抑制特殊的腐败微生物繁殖具有非常重要的意义。Ercolini等 [31]调查了将相同的牛肉在露天放置、非真空包装、真空包装以及抗菌包装4 种包装条件下放入4 ℃贮藏45 d过程中腐败微生物的变化规律。结果显示,热杀索丝菌在露天放置和非真空包装的初期均为优势类群,在露天放置的贮藏后期优势类群被假单胞菌所取代,而非真空包装的贮藏后期腐败微生物却以广布肉杆菌为主。在真空包装贮藏的后期,LAB占据主要优势地位,而与此同时抗菌包装的牛肉中95%的细菌序列被鉴定为广布肉杆菌。
食品污染来源是食品微生物生态学研究的热点和难点。评价食品相关环境中的微生物群落特征对于认识食品污染来源起着至关重要的作用。通常,原料来源是食品污染最重要的来源,原料所携带的微生物同时也影响着后期食品产品的风味特征 [63-64]。de Filippis等 [32]研究牛排污染腐败的微生物来源,发现引起牛排污染的主要微生物假单胞菌和热杀索丝菌均在牛肉来源的动物残骸上被检测到,并且同一块残骸上不同的切口也对牛排的污染程度具有一定的影响。最近,Bokulich等 [56]调查了酿酒葡萄表面的微生物生物地理分布,指出葡萄采摘方式、气候条件、地理条件和葡萄品种共同影响着葡萄表面的微生物区系,而不同地区葡萄表面微生物的差异又显著影响了后期葡萄酒发酵过程,导致了各地葡萄酒风味的各不相同。葡萄表面微生物的这种“地缘特征”最终塑造了不同地区葡萄酒风味的“地域风味”。
食源性病原菌是食品生产、发酵和贮藏过程中必须重点关注的检测指标。因此各种针对特殊病原菌设计的特异引物被广泛用于食品中病原菌的PCR分子诊断 [65-67]。但是,为了避免假阳性的出现,必须对PCR扩增产物进行验证。高通量测序具有高度的灵敏性,因此该技术可用于该类研究应用中。目前,高通量测序已成功用于弯曲杆菌特异性PCR分析的验证中,指出特异性PCR分析不适于微生物区系复杂的自然环境中病原菌的检测 [68]。与病原菌检测类似,对一些重要的有益微生物类群进行特异性检测也具有重要意义。最近,de Filippis等 [39]使用lacS基因特异性引物LCS62f(5’-GGCTTCCAATACTTTAATT-3’)和LCS312r(5’-AAGTGAGTTGTCACAAACAT-3’)对奶酪制品中嗜热链球菌进行检测,高通量测序结果表明不同样品来源的嗜热链球菌lacS基因存在差异,共识别出28 种不同的lacS基因类型,揭示非rRNA基因标记也可能适用于微生物生物型的定量监测中。
对食品微生物生态学的研究主要集中在细菌和真菌的研究中,而对古菌的研究则相对有限。最近的一项研究表明,发酵海鲜中古菌的多样性显著高于细菌,这与其他环境的研究结果刚好相反,表明在发酵海鲜食品的发酵过程中,古菌可能扮演了主要角色 [47]。焦磷酸测序结果显示,发酵海鲜食品中古菌主要属于极端嗜盐古菌的盐杆菌科和中温泉古菌的泉古菌门群I.1、深海群B、杂泉古菌门群3 个系统发育类群。到目前为止,这些中温泉古菌类群鲜见于其他发酵食品。
高通量测序在食品微生物生态学研究中应用,极大加深了人们对食品微生物多样性的认识。到目前为止,鲜有一种免培养分析方法能获得像高通量测序如此大的数据量,因此,高通量测序能快速、准确获得食品中大多数的微生物信息,描述微生物群落结构的演替规律(表2)。同时,高通量测序能同时分析多个食品样品中的微生物区系,极大地缩短了分析时间,并减少了因多次实验造成的误差。此外,高通量测序减少了许多有毒试剂的使用,与其他分子生物学方法相比更加安全,而且高通量测序主要为仪器自动完成,能在很大程度上节省实验者的工作时间。
表2 高通量测序研究食品微生物生态学的优势和局限性
Table 2 Advantages and disadvantages of high-throughput sequencing in food microbial ecology

优势局限性可同时分析多个食品样品测序长度短,限制了在种水平的分析安全性和自动化程度高数据生物信息学分析难度大可靠性高,误差小测序费用较高数据量大,分析全面
与其他免培养分析方法一样,高通量测序也存在不足之处(表2)。高通量测序最大的不足之处在于其数据量巨大,对数据的处理和解析相当繁琐,需要专业的分析平台和熟练的生物信息学分析能力,因此限制了其在食品生产中的应用。同时,与其他分子生物学技术相比,高通量测序成本较高,进一步限制了其在食品工业中的推广。此外,高通量测序结果的序列读长有限(1 000 bp以下),对序列进行鉴定时通常只能到属水平,而在食品生产检测中,更希望能在种一级的水平上对微生物区系进行研究。也正是因为读长的限制,引物的不当选择也可能导致对食品中微生物多样性的高估 [22]。
近年来,高通量测序技术被广泛应用于食品微生物分子生态学研究,目前已在分析食品微生物多样性、评价食品发酵和贮藏过程中微生物群落结构变化、监测食源性病原菌动态特征以及控制食品质量等方面发挥出无可比拟的优势。高通量测序在食品微生物基因组研究中的应用更有利于食源性病原菌基因组的快速分析 [69]。在进一步的研究中,可将高通量测序用于食品微生物转录组学研究,从群体水平研究食品微生物功能基因的表达水平及其在不同条件下的转录调控机制。此外,应在后续的研究中对生物信息学分析工具进行改进,使海量数据分析更加简便,使高通量测序更加广泛地应用于食品微生物生态学的研究。
参考文献:
[1] AMPE F, SIRVENT A, ZAKHIA N. Dynamics of the microbial community responsible for traditional sour cassava starch fermentation studied by denaturing gradient gel electrophoresis and quantitative rRNA hybridization[J]. International Journal of Food Microbiology, 2001, 65(1): 45-54. DOI:10.1016/S0168-1605(00)00502-X.
[2] 董玲, 蒲彪, 敖晓琳, 等. 四川冬菜中细菌群落组成及多样性[J]. 微生物学报, 2012, 52(4): 519-525.
[3] 蓝蔚青, 谢晶, 周会, 等. 不同时期鲳鱼冷藏期间优势腐败菌的多样性变化[J]. 食品科学, 2015, 36(2): 226-231. DOI:10.7506/spkx1002-6630-201502044.
[4] 舒畅, 吴春生, 钟慈平, 等. 发酵食品微生物多样性研究方法进展[J].食品科学, 2013, 34(15): 397-402. DOI:10.7506/spkx1002-6630-201315076.
[5] 高蕙文, 吕欣, 董明盛. PCR-DGGE指纹技术在食品微生物研究中的应用[J]. 食品科学, 2005, 26(8): 465-468.
[6] BOKULICH N A, BAMFORTH C W, MILLS D A. Brewhouseresident microbiota are responsible for multi-stage fermentation of American coolship ale[J]. PLoS ONE, 2012, 7(4): e35507. DOI:10.1371/journal.pone.0035507.
[7] MASOUD W, VOGENSEN F K, LILLEVANG S, et al. The fate of indigenous microbiota, starter cultures, Escherichia coli, Listeria innocua and Staphylococcus aureus in Danish raw milk and cheeses determined by pyrosequencing and quantitative real time (qRT)-PCR[J]. International Journal of Food Microbiology, 2012, 153(1): 192-202. DOI:10.1016/j.ijfoodmicro.2011.11.014.
[8] ERCOLINI D. High-throughput sequencing and metagenomics: moving forward in the culture-independent analysis of food microbial ecology[J]. Applied and Environmental Microbiology, 2013, 79(10): 3148-3155. DOI:10.1128/AEM.00256-13.
[9] SOLIERI L, DAKAL T C, GIUDICI P. Next-generation sequencing and its potential impact on food microbial genomics[J]. Annals of Microbiology, 2013, 63(1): 21-37. DOI:10.1007/s13213-012-0478-8.
[10] LINDAHL B D, NILSSON R H, TEDERSOO L, et al. Fungal community analysis by high-throughput sequencing of amplified markers-a user's guide[J]. New Phytologist, 2013, 199(1): 288-299. DOI:10.1111/nph.12243.
[11] 岳桂东, 高强, 罗龙海, 等. 高通量测序技术在动植物研究领域中的应用[J]. 中国科学(生命科学), 2012, 42(2): 107-124. DOI:10.1360/052011-634.
[12] METZKER M L. Sequencing technologies-the next generation[J]. Nature Reviews Genetics, 2010, 11(1): 31-46. DOI:10.1038/nrg2626.
[13] LOMAN N J, MISRA R V, DALLMAN T J, et al. Performance comparison of benchtop high-throughput sequencing platforms[J]. Nature Biotechnology, 2012, 30(5): 434-439. DOI:10.1038/nbt.2198.
[14] LUO C, TSEMENTZI D, KYRPIDES N, et al. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample[J]. PLoS ONE, 2012, 7(2): e30087. DOI:10.1371/journal.pone.0030087.
[15] ROESCH L F W, FULTHOTPE R R, RIVA A, et al. Pyrosequencing enumerates and contrasts soil microbial diversity[J]. The ISME Journal, 2007, 1(4): 283-290. DOI:10.1038/ismej.2007.53.
[16] QIU M, ZHANG R, XUE C, et al. Application of bio-organic fertilizer can control Fusarium wilt of cucumber plants by regulating microbial community of rhizosphere soil[J]. Biology and Fertility of Soils, 2012, 48(7): 807-816. DOI:10.1007/s00374-012-0675-4.
[17] ZIMMERMAN N B, VITOUSEK P M. Fungal endophyte communities reflect environmental structuring across a Hawaiian landscape[J]. Proceedings of the National Academy of Sciences USA, 2012, 109(32): 13022-13027. DOI:10.1073/pnas.1209872109.
[18] SONG Z Q, WANG F P, ZHI X Y, et al. Bacterial and archaeal diversities in Yunnan and Tibetan hot springs, China[J]. Environmental Microbiology, 2013, 15(4): 1160-1175. DOI:10.1111/1462-2920.12025.
[19] 郑艺, 张家超, 郭壮, 等. 基于高通量测序技术分析肠道菌群及其影响因素的研究进展[J]. 中国食品学报, 2014, 14(11): 157-164.
[20] 王小芬, 王伟东, 高丽娟, 等. 变性梯度凝胶电泳在环境微生物研究中的应用详解[J]. 中国农业大学学报, 2006, 11(5): 1-7.
[21] QUIGLEY L, O’SULLIVAN O, BERESFORDT P, et al. A comparison of methods used to extract bacterial DNA from raw milk and raw milk cheese[J]. Journal of Applied Microbiology, 2012, 113(1): 96-105. DOI:10.1111/j.1365-2672.2012.05294.x
[22] SUN D L, JIANG X, WU Q L, et al. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity[J]. Applied and Environmental Microbiology, 2013, 79(19): 5962-5969. DOI:10.1128/AEM.01282-13.
[23] HAAS B J, GEVERS D, EARL A M, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J]. Genome Research, 2011, 21(3): 494-504. DOI:10.1101/gr.112730.110.
[24] QUAIL M A, SMITH M, COUPLAND P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers[J]. BMC Genomics, 2012, 13(1): 341. DOI:10.1186/1471-2164-13-341.
[25] LEITH A M O, MAYO B, RACHID C, et al. Assessment of the microbial diversity of Brazilian kefir grains by PCR-DGGE and pyrosequencing analysis[J]. Food Microbiology, 2012, 31(2): 215-221. DOI:10.1016/j.fm.2012.03.011.
[26] KIYOHARA M, KOYANAGI T, MATSUI H, et al. Changes in microbiota population during fermentation of narezushi as revealed by pyrosequencing analysis[J]. Bioscience, Biotechnology, and Biochemistry, 2012, 76(1): 48-52. DOI:10.1271/bbb.110424.
[27] NAM Y D, PARK S, LIM S I. Microbial composition of the Korean traditional food “kochujang” analyzed by a massive sequencing technique[J]. Journal of Food Science, 2012, 77(4): M250-M256. DOI:10.1111/j.1750-3841.2012.02656.x.
[28] NAM Y D, YI S H, LIM S I. Bacterial diversity of cheonggukjang, a traditional Korean fermented food, analyzed by barcoded pyrosequencing[J]. Food Control, 2012, 28(1): 135-142. DOI:10.1016/ j.foodcont.2012.04.028.
[29] NAM Y D, LEE S Y, LIM S I. Microbial community analysis of Korean soybean pastes by next-generation sequencing[J]. International Journal of Food Microbiology, 2012, 155(1): 36-42. DOI:10.1016/ j.ijfoodmicro.2012.01.013.
[30] REINA L D, P REZ-D AZ I M, BREIDT F, et al. Characterization of the microbial diversity in yacon spontaneous fermentation at 20 ℃[J]. International Journal of Food Microbiology, 2015, 203: 35-40. DOI:10.1016/j.ijfoodmicro.2015.03.007.
[31] ERCOLINI D, FERROCINO I, NASI A, et al. Monitoring of microbial metabolites and bacterial diversity in beef stored under different packaging conditions[J]. Applied and Environmental Microbiology, 2011, 77(20): 7372-7381. DOI:10.1128/AEM.05521-11.
[32] de FILIPPIS F, la STORIA A, VILLANI F, et al. Exploring the sources of bacterial spoilers in beefsteaks by culture-independent high-throughput sequencing[J]. PLoS ONE, 2013, 8(7): e70222. DOI:10.1371/journal.pone.0070222.
[33] del RBOL J T, PULIDO R P, la STORIA A, et al. Changes in microbial diversity of brined green asparagus upon treatment with high hydrostatic pressure[J]. International Journal of Food Microbiology, 2016, 216: 1-8. DOI:10.1016/j.ijfoodmicro.2015.09.001.
[34] COCOLIN L, ALESSANDRIA V, BOTTA C, et al. NaOH-debittering induces changes in bacterial ecology during table olives fermentation[J]. PLoS ONE, 2013, 8(7): e69074. DOI:10.1371/journal. pone.0069074.
[35] GREPPI A, FERROCINO I, la STORIA A, et al. Monitoring of the microbiota of fermented sausages by culture independent rRNA-based approaches[J]. International Journal of Food Microbiology, 2015, 212: 67-75. DOI:10.1016/j.ijfoodmicro.2015.01.016.
[36] KIM Y S, KIM M C, KWON S W, et al. Analyses of bacterial communities in meju, a Korean traditional fermented soybean bricks, by cultivation-based and pyrosequencing methods[J]. The Journal of Microbiology, 2011, 49(3): 340-348. DOI:10.1007/s12275-011-0302-3.
[37] PARK E J, CHUN J, CHA C J, et al. Bacterial community analysis during fermentation of ten representative kinds of kimchi with barcoded pyrosequencing[J]. Food Microbiology, 2012, 30(1): 197-204. DOI:10.1016/j.fm.2011.10.011.
[38] GUIDONE A, ZOTTA T, MATERA A, et al. The microbiota of highmoisture mozzarella cheese produced with different acidification methods[J]. International Journal of Food Microbiology, 2016, 216: 9-17. DOI:10.1016/j.ijfoodmicro.2015.09.002.
[39] de FILIPPIS F, la STORIA A, STELLATO G, et al. A selected core microbiome drives the early stages of three popular Italian cheese manufactures[J]. PLoS ONE, 2014, 9(2): e89680. DOI:10.1371/ journal.pone.0089680.
[40] JUNG M J, NAM Y D, ROH S W, et al. Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters[J]. Food Microbiology, 2012, 30(1): 112-123. DOI:10.1016/ j.fm.2011.09.008.
[41] GAROFALO C, OSIMANI A, MILANOVIĆ V, et al. Bacteria and yeast microbiota in milk kefir grains from different Italian regions[J]. Food Microbiology, 2015, 49(1): 123-133. DOI:10.1016/ j.fm.2015.01.017.
[42] LIU W, ZHENG Y, KWOKL Y, et al. High-throughput sequencing for the detection of the bacterial and fungal diversity in Mongolian naturally fermented cow’s milk in Russia[J]. BMC Microbiology, 2015, 15(1): 385. DOI:10.1186/s12866-015-0385-9.
[43] ALDRETE-TAPIA A, ESCOBAR-RAM REZ M C, TAMPLIN M L, et al. High-throughput sequencing of microbial communities in Poro cheese, an artisanal Mexican cheese[J]. Food Microbiology, 2014, 44: 136-141. DOI:10.1016/j.fm.2014.05.022.
[44] ERCOLINI D, PONTONIO E, de FILIPPIS F, et al. Microbial ecology dynamics during rye and wheat sourdough preparation[J]. Applied and Environmental Microbiology, 2013, 79(24): 7827-7836. DOI:10.1128/ AEM.02955-13.
[45] LIU W, XI X, SUDU Q, et al. High-throughput sequencing reveals microbial community diversity of Tibetan naturally fermented yak milk[J]. Annals of Microbiology, 2015, 65(3): 1741-1751. DOI:10.1007/s13213-014-1013-x.
[46] ERCOLINI D, de FILIPPIS F, LA STORIA A, et al. “Remake”by high-throughput sequencing of the microbiota involved in the production of water buffalo mozzarella cheese[J]. Applied and Environmental Microbiology, 2012, 78(22): 8142-8145. DOI:10.1128/ AEM.02218-12.
[47] ROH S W, KIM K H, NAM Y D, et al. Investigation of archaeal and bacterial diversity in fermented seafood using barcoded pyrosequencing[J]. The ISME Journal, 2010, 4(1): 1-16. DOI:10.1038/ ismej.2009.83.
[48] MASOUD W, TAKAMIYA M, VOGENSEN F K, et al. Characterization of bacterial populations in Danish raw milk cheeses made with different starter cultures by denaturating gradient gel electrophoresis and pyrosequencing[J]. International Dairy Journal, 2011, 21(3): 142-148. DOI:10.1016/j.idairyj.2010.10.007.
[49] POLKA J, REBECCHI A, PISACANE V, et al. Bacterial diversity in typical Italian salami at different ripening stages as revealed by high-throughput sequencing of 16S rRNA amplicons[J]. Food Microbiology, 2015, 46: 342-356. DOI:10.1016/j.fm.2014.08.023.
[50] RIQUELME C, C MARA S, de LURDES M N, et al. Characterization of the bacterial biodiversity in Pico cheese (an artisanal Azorean food)[J]. International Journal of Food Microbiology, 2015, 192: 86-94. DOI:10.1016/j.ijfoodmicro.2014.09.031.
[51] REBECCHI A, PISACANE V, MIRAGOLI F, et al. High-throughput assessment of bacterial ecology in hog, cow and ovine casings used in sausages production[J]. International Journal of Food Microbiology, 2015, 212: 49-59. DOI:10.1016/j.ijfoodmicro.2015.04.047.
[52] BOKULICH N A, JOSEPH C M, ALLEN G, et al. Next-generation sequencing reveals significant bacterial diversity of botrytized wine[J]. PLoS ONE, 2012, 7(5): e36357. DOI:10.1371/journal.pone.0036357.
[53] BOKULICH N A, AMIRANASHVILI L, CHITCHYAN K, et al. Microbial biogeography of the transnational fermented milk matsoni[J]. Food Microbiology, 2015, 50: 12-19. DOI:10.1016/ j.fm.2015.01.018.
[54] QUIGLEY L, O’SULLIVAN O, BERESFORD T P, et al. Highthroughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses[J]. Applied and Environmental Microbiology, 2012, 78(16): 5717-5723. DOI:10.1128/ AEM.00918-12.
[55] DOBSON A, O’SULLIVAN O, COTTER P D, et al. High-throughput sequence-based analysis of the bacterial composition of kefir and an associated kefir grain[J]. FEMS Microbiology Letters, 2011, 320(1): 56-62. DOI:10.1111/j.1574-6968.2011.02290.x.
[56] BOKULICH N A, THORNGATE J H, RICHARDSON P M, et al. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate[J]. Proceedings of the National Academy of Sciences USA, 2014, 111(1): 139-148. DOI:10.1073/ pnas.1317377110.
[57] MARSH A J, O’SULLIVAN O, HILL C, et al. Sequencing-based analysis of the bacterial and fungal composition of kefir grains and milks from multiple sources[J]. PLoS ONE, 2013, 8(7): e69371. DOI:10.1371/journal.pone.0069371.
[58] MARSH A J, O’SULLIVAN O, HILL C, et al. Sequence-based analysis of the bacterial and fungal compositions of multiple kombucha (tea fungus) samples[J]. Food Microbiology, 2014, 38: 171-178. DOI:10.1016/j.fm.2013.09.003.
[59] MARSH A J, O’SULLIVAN O, HILL C, et al. Sequence-based analysis of the microbial composition of water kefir from multiple sources[J]. FEMS Microbiology Letters, 2013, 348(1): 79-85. DOI:10.1111/1574-6968.12248.
[60] ALEGR A , SZCZESNY P, MAYO B, et al. Biodiversity in Oscypek, a traditional Polish cheese, determined by culturedependent and-independent approaches[J]. Applied and Environmental Microbiology, 2012, 78(6): 1890-1898. DOI:10.1128/AEM.06081-11.
[61] SAKAMOTO N, TANAKA S, SONOMOTO K, et al. 16S rRNA pyrosequencing-based investigation of the bacterial community in nukadoko, a pickling bed of fermented rice bran[J]. International Journal of Food Microbiology, 2011, 144(3): 352-359. DOI:10.1016/ j.ijfoodmicro.2010.10.017.
[62] ERCOLINI D, HILL P J, DODD C E R. Bacterial community structure and location in Stilton cheese[J]. Applied and Environmental Microbiology, 2003, 69(6): 3540-3548. DOI:10.1128/AEM.69.6.3540-3548.2003.
[63] BOKULICH N A, MILLS D A. Facility-specific “house” microbiome drives microbial landscapes of artisan cheesemaking plants[J]. Applied and Environmental Microbiology, 2013, 79(17): 5214-5223. DOI:10.1128/AEM.00934-13.
[64] CRUCIATA M, SANNINO C, ERCOLINI D, et al. Animal rennets as sources of dairy lactic acid bacteria[J]. Applied and Environmental Microbiology, 2014, 80(7): 2050-2061. DOI:10.1128/AEM.03837-13.
[65] POSTOLLEC F, FALENTIN H, PAVAN S, et al. Recent advances in quantitative PCR (qPCR) applications in food microbiology[J]. Food Microbiology, 2011, 28(5): 848-861. DOI:10.1016/j.fm.2011.02.008.
[66] DWIVEDI H P, JAYKUS L A. Detection of pathogens in foods: the current state-of-the-art and future directions[J]. Critical Reviews in Microbiology, 2011, 37(1): 40-63. DOI:10.3109/104084 1X.2010.506430.
[67] REMENANT B, JAFFR S E, DOUSSET X, et al. Bacterial spoilers of food: behavior, fitness and functional properties[J]. Food Microbiology, 2015, 45(PtA): 45-53. DOI:10.1016/j.fm.2014.03.009.
[68] OAKLEY B B, LINE J E, BERRANG M E, et al. Pyrosequencingbased validation of a simple cell-suspension polymerase chain reaction assay for Campylobacter with application of high-processivity polymerase and novel internal amplification controls for rapid and specific detection[J]. Diagnostic Microbiology and Infectious Disease, 2012, 72(2): 131-138. DOI:10.1016/j.diagmicrobio.2011.11.001.
[69] GILMOUR M W, GRAHAM M, van DOMSELAAR G, et al. Highthroughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak[J]. BMC Genomics, 2010, 11(1): 120. DOI:10.1186/1471-2164-11-120.
Application of High-Throughput Sequencing in Food Microbial Ecology: A Review
MI Qili
1,2, LI Xuemei
1, GUAN Ying
1, GAO Qian
1, GUI Yongfa
1, ZHU Zhouhai
1, YAO Jianhua
1,*
(1. Technology Center of China Tobacco Yunnan Industrial Co. Ltd., Kunming 650106, China; 2. Yunnan Institute of Microbiology, Yunnan University, Kunming 650091, China)
Abstract:With the development of molecular biology, a growing number of new strategies have been used in studies in food microbiology. High-throughput sequencing, a newly burgeoning culture-independent technique, is characterized by rapid, sensitive and comprehensive analysis of large volume of data and has been recently introduced into food microbial ecology. In this review, we summarize the process of high-throughput sequencing and its application in food microbial ecology. The limitations and advantages of this technique are discussed, and future prospects for its application in food microbial ecology are presented as well. The aim is to provide a reference for the development of food microbial ecology.
Key words:food microbial ecology; high-throughput sequencing; diversity; microf ora
DOI:10.7506/spkx1002-6630-201623049
中图分类号:Q938
文献标志码:A
文章编号:
引文格式:
米其利, 李雪梅, 管莹, 等. 高通量测序在食品微生物生态学研究中的应用[J]. 食品科学, 2016, 37(23): 302-308.
DOI:10.7506/spkx1002-6630-201623049. http://www.spkx.net.cn
MI Qili, LI Xuemei, GUAN Ying, et al. Application of high-throughput sequencing in food microbial ecology: a review[J]. Food Science, 2016, 37(23): 302-308. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201623049. http://www.spkx.net.cn
收稿日期:2015-10-22
基金项目:云南中烟工业有限责任公司项目(2015JC01;2015JC03);中国烟草总公司项目(110201502006)
作者简介:米其利(1981—),女,助理研究员,博士研究生,主要从事微生物资源与应用研究。E-mail:miqiliyn@126.com
*通信作者:夭建华(1968—),女,研究员,博士,主要从事烟草微生物资源与应用研究。E-mail:jhyao_2007@126.com