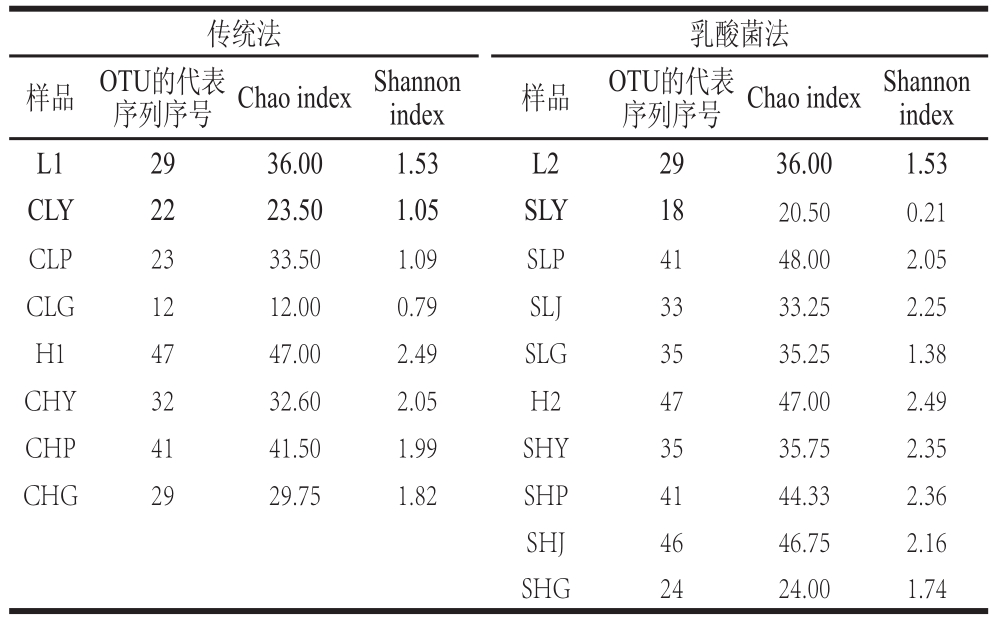
表1 腌干鱼加工过程-多样性
Table1 -Diversity measurement for dried-salted fish from different processing stages
吴燕燕1,钱茜茜1,2,李来好1,陈胜军1,邓建朝1,李春生1
(1.中国水产科学研究院南海水产研究所,农业部水产品加工重点实验室,广东 广州 510300;2.上海海洋大学食品学院,上海 201306)
摘 要:为研究传统和乳酸菌法加工腌干鱼过程微生物的变化规律,为优化腌干鱼工艺提供理论依据,采用MiSeq测序技术,研究腌干鱼在不同加工阶段的细菌多样性,结果表明:腌干鱼加工过程微生物种类丰富,主要分为三大类:拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和变性菌门(Proteobacteria);蓝圆鲹初始菌相中优势细菌为肠杆菌科(Enterbacteriaceae)、肠球菌科(Enterococcactae)、假单胞菌科(Pseudomonadaceae)和希瓦氏菌科(Shewanellaceae);海鲈初始菌相中优势菌主要是气单胞菌科(Aeromonadaceae)、芽孢杆菌科(Bacillaceae)、葡萄球菌科(Staphylococcaceae)、丛毛单胞菌科(Comamonadaceae)、肠杆菌科(Enterbacteriaceae)、肠球菌科(Enterococcaceae)、摩式摩根菌科(Moraganellaceae)、链球菌科(St reptococcaceae)等。传统腌制加工过程中菌相比较单一,优势菌主要是弧菌科(Vibrionaceae)、葡萄球菌科(Staphylococcaceae)、假单胞菌科(Pseudomonadaceae)和动性球菌科(Planococcaceae);而接种乳酸菌发酵后,加工过程中细菌多样性呈现增加趋势,并且乳酸菌成为优势菌,促进了微杆菌科(Exiguobacteracea),葡萄球菌(Staphylococcaceae)等优势菌的繁殖;此外还检测到气单胞菌(Aeromonadaceae)和乳杆菌(Lactobacillaceae)等传统蓝圆鲹腌制加工过程中没有出现的菌。海鲈在腌制加工过程中细菌多样性明显高于蓝圆鲹,乳酸菌法腌制加工过程中细菌多样性明显增加。
关键词:腌干鱼;Illumina MiSeq测序;微生物多样性;优势菌
传统腌干鱼制品由于其易保存、营养丰富、风味独特,深受人们喜爱。腌干鱼在腌制加工过程中伴随着极为复杂的理化变化和微生物变化[1]。多年来,国内外学者对腊鱼[2]、咸鱼[3-4]、plaa-som[5]等鱼制品进行菌相分析,发现乳酸菌、葡萄球菌和微球菌是优势菌群。腌制食品的加工一般要经过腌制和干制两道工序,腌制主要是食盐渗透的过程,对微生物进行选择性培养,杀死或抑制一部分腐败菌的生长繁殖;干制则是脱去大部分水分,干制过程中有大量的乳酸菌、微球菌和葡萄球菌生长。传统的腌干鱼制品主要通过自然发酵生成,但是这种发酵工艺已不适合现代化生产的要求。这是由于传统腌干鱼肉中的微生物包括鱼肉本身自带的微生物和环境中混有的杂菌,随着环境和加工条件的变化,容易导致发酵过程中的微生物群落结构不稳定,影响产品的风味和质量[6]。因此,人们越来越倾向于应用有益微生物来实现发酵过程的有效控制,对微生物群落结构进行有效调控,并缩短腌制时间,改善产品品质,保证产品的食用安全性[7-9]。
长期以来,微生物群落多样性研究主要还是基于传统的分离、纯化以及通过一系列繁琐的生理生化实验进行鉴定,这种方法不仅耗时耗力,而且不能对分离物进行精确鉴定,无法获得微生物群落多样性的真正概貌[10]。因为有些微生物只能在特定的环境中生长,有些甚至不可培养[11]。Handelsman等[12]首次提出宏基因组概念:指在特定环境样品中基因组的总和,包含细菌基因组和真菌基因组。宏基因组学采用新一代的高通量测序技术(Illumina MiSeq)直接对样品中微生物总DNA进行测序,不需要对微生物进行分离培养,不仅可以检测到低丰度的微生物,而且速度快,结果准确。聂志强等[12]采用宏基因组技术对天津独流老醋醋酸发酵过程中细菌群落组成及其多样性进行了分析,结果清晰地揭示了食醋这一传统发酵食品酿造过程中丰富的微生物多样性以及与代谢产物之间的联系。除此之外,国内外的诸多学者也用现代分子生物学手段清晰地揭示了微生物多样性变化[13-16]。
本研究从腌干鱼加工过程中的几个关键环节取样,应用Illumina MiSeq测序技术分析样品中的微生物群落结构以及随着加工的进行微生物群落的变化。探讨添加乳酸菌发酵剂后鱼肉中微生物群落的结构变化以及发酵剂在腌干鱼加工过程中能否成为优势菌,这为优化腌干鱼的生产工艺提供参考依据。
1.1 材料与试剂
蓝圆鲹(Decapterus maruadsi),俗名池鱼、巴浪鱼,体长30~35 cm,体质量200~220 g,冰鲜;海鲈鱼(Lateolabrax japonicus),俗名日本真鲈、七星鲈,体长35~40 cm,500~600 g),鲜活,均为市购。
细菌基因组DNA提取试剂盒 美国Omega科技公司。
1.2 仪器与设备
高速冷冻离心机 美国Bio-Rad公司;MiSeq PE250美国Illumina公司;GeneJET胶回收试剂盒 美国Thermo Scientific公司;引物515F和806R由深圳华大基因有限公司合成;HH-4数显恒温水浴锅 常州澳华仪器有限公司;MM-2快速涡旋混合器 姜堰市沈高康健生化器具厂。
1.3 方法
1.3.1 样品制备
腌干鱼的制作工艺参照参考文献[5],调整腌制时间,蓝圆鰺腌制48 h,海鲈腌制72 h。取传统腌干鱼加工过程中4 个阶段产品的背部肌肉部分作为试材,每阶段抽2 条鱼,装入无菌自封袋中于-20 ℃冰箱保存待测。其中,传统法腌干蓝圆鲹的4 个阶段分别以L1、CLY2、CLP、CLG2表示,合称为A组。L1:新鲜蓝圆鲹;CLY2:腌制48 h后的样品;CLP:浸泡脱盐后的样品;CLG2:烘干成品。传统法腌干海鲈的4 个阶段分别以H1、CHY、CHP、CHG表示,合称为C组。H1:新鲜海鲈鱼;CHY:腌制72 h后的样品;CHP:浸泡脱盐后的样品;CHG:烘干成品。
取乳酸菌法腌干鱼加工过程中5 个阶段产品的背部肌肉部分作为试材,每阶段抽2 条鱼,装入无菌自封袋中于-20 ℃冰箱保存待测。其中,乳酸菌法腌干蓝圆鲹的5 个阶段分别以L2、SLY、SLP、SLJ、SLG表示,合称为B组。L2:新鲜蓝圆鲹;SLY:腌制24 h后的样品;SLP:浸泡脱盐后的样品;SLJ:接种发酵17 h后的样品;SLG:烘干成品。乳酸菌法腌干海鲈5 个阶段分别以H2、SHY、SHP、SHJ、SHG表示,合称为D组。H2:新鲜海鲈鱼;SHY:腌制24 h后的样品;SHP:浸泡脱盐后的样品;SHJ:接种发酵17 h后的样品;SHG:烘干成品。
1.3.2 细菌总DNA的提取
取20 g剪碎样品,置于300 mL的锥形瓶里,加入220 mL无菌生理盐水,振荡混匀,取1 mL混合液于装有9 mL灭菌LB肉汤培养基的试管中富集培养24 h。采用美国Omega科技有限公司的提取试剂盒(DP302)提取细菌基因组DNA,提取步骤参照试剂盒产品说明书。
1.3.3 Illumina MiSeq测序
研究表明16S rDNA高变区序列所得到的物种信息与其全长序列结果差异不大,且高变区片段更短,在进行大量的数据分析时不仅能够提高研究效率,而且可以降低成本[17]。故设计细菌的16S rDNA序列V4片段扩增的通用引物,正向引物选用515F(5’-GTGCCAGCMGCCGCGGTAA-3’),反向引物选用806R(5’-GGACTACNNGGG TATCTAAT-3’)。PCR反应体系:4 μL Primer Cocktail,25 μL Master Mix,2 μL DNA和19 μL ddH2O。反应参数:98 ℃预变性3 min;98 ℃变性45 s,55 ℃退火45 s,72 ℃延伸45 s,30 个循环;72 ℃延伸7 min。
PCR扩增产物使用1.0%琼脂糖凝胶电泳检测,等浓度混样,使用Thermo Scientific公司的GeneJET胶回收试剂盒回收纯化产物,建库(NEB Next®Ultra™ DNA Library Prep Kit for Illumina)并于Illumina MiSeq PE250(PE251+8+251)上机测序。
1.4 数据分析
根据Barcode将下机数据(raw data)拆分为不同样品数据,并截去Barcode序列和PCR扩增引物序列。使用FLASH[18](Fast Length Adjustment of Short reads,v1.2.11),将reads拼接成Raw Tags,利用重叠关系将双末端测序得到的成对reads组装成一条序列,得到高变区的Tags[19],将经上述处理后的Tags与Gold database(v20110519)进行比对(UCHIME Algorithm)检测嵌合体序列[20],除去嵌合体序列得到Effective Tags。用UPARSE在97%相似度条件下进行聚类,得到OTU的代表序列(OTUs)。得到OTU代表序列后,通过RDP classifer(v2.2)软件将OTU代表序列与数据库有比对进行物种注释[21],置信度阈值设置为0.8,并用R(v3.1.1)软件绘制主成分分析(principal component analysis,PCA)图。同时计算α-多样性,包括物种观察指数(observed species index)、赵氏指数(Chao index)、艾斯指数(ACE index)、香农指数(Shannon index)及辛普森指数(Simpson index),用稀释曲线评价测序深度[22]。
2.1 α-多样性分析
α-多样性反映了单个样品内部物种的多样性,Chao index反映样品中群落的丰富度,即简单指群落中物种的数量,而不考虑群落中每个物种的丰度情况,Shannon index反映群落的多样性,受样品群落中物种丰富度和物种均匀度的影响。
表1 腌干鱼加工过程-多样性
Table1 -Diversity measurement for dried-salted fish from different processing stages
经过拼接,过滤等[23]一系列处理,蓝圆鲹在传统腌制加工过程中共获得104 474 条细菌序列,在乳酸菌辅助腌制加工过程中共获得131 434 条细菌系列;海鲈在传统腌制加工过程中共获得细菌序列105 289 条,在乳酸菌辅助腌制加工过程中共获得细菌序列131 745 条。从这些数据可以看出乳酸菌辅助腌制加工方法中细菌多样性比传统法丰富,海鲈腌制加工过程中的细菌多样性略高于蓝圆鲹。不同加工阶段的样品微生物多样性指数,如表1所示,腌干鱼在腌制阶段微生物多样性呈下降趋势,由于高浓度食盐的渗入以及发酵产生的低pH值杀死了部分微生物,到漂洗阶段,鱼肉充分暴露在空气中使得细菌多样性增加,干燥阶段随着水分活度的降低,微生物多样性也相应降低。总体上细菌物种多样性均呈现下降趋势。图1的样品稀释曲线也展示了这一趋势。图1曲线均已达到平坦期,即测序数据量合理,测序深度已基本覆盖样品中所有微生物。
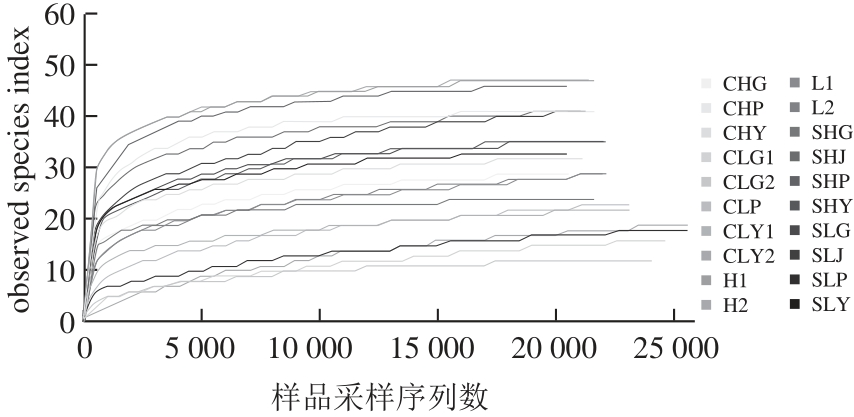
图1 稀释曲线
Fig.1 Rarefaction curve
图2为A、B、C、D组间α-多样性盒形图,更直观显示组间α-多样性差异。observed species index、Chao index、ACE index反映样品中群落的丰富度;Shannon index以及Simpson index反映群落的多样性。Simpson index越小,代表样品中的物种越丰富,从表1可以看出,海鲈鱼中的微生物多样性明显比蓝圆鰺丰富,加入乳酸菌发酵剂之后,促进了优势菌的生长繁殖,因此也丰富了物种多样性,所以海鲈鱼多样性高于蓝圆鰺,乳酸菌法高于传统法。从图2 α-多样性盒形图也可以看出,与其他3 组相比,D组的observed species index、Chao index、ACE index和Shannon index均较高,而Simpson index较低。因此,D组样品具有最高的细菌多样性,其次为C组>B组>A组。
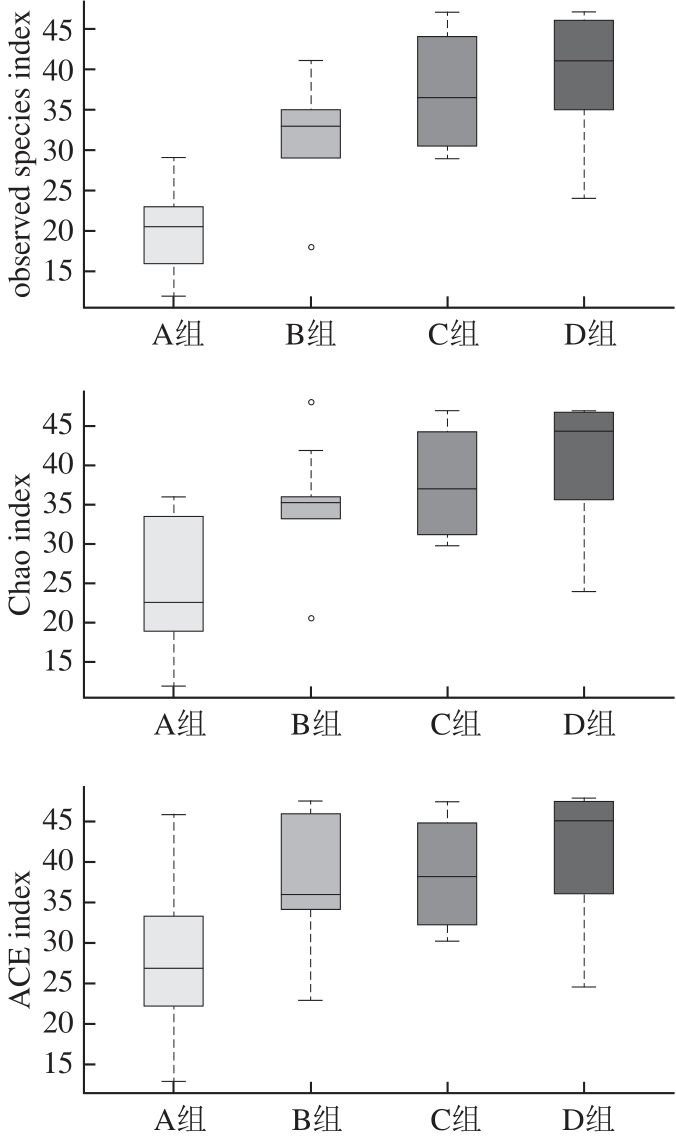
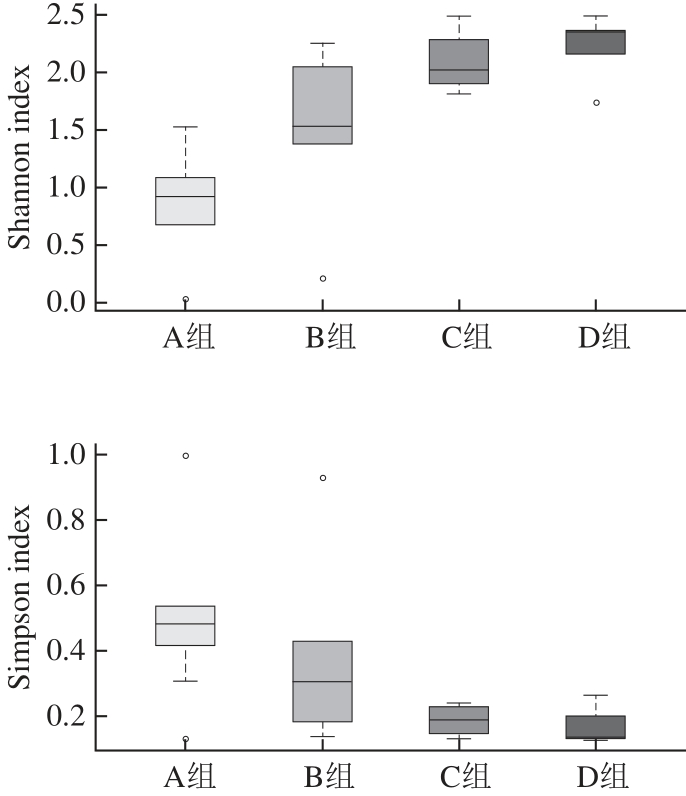
图2 2 α-多样性指数的箱图
Fig.2 Boxplots of α-diversity indexes
2.2 OUT PCA
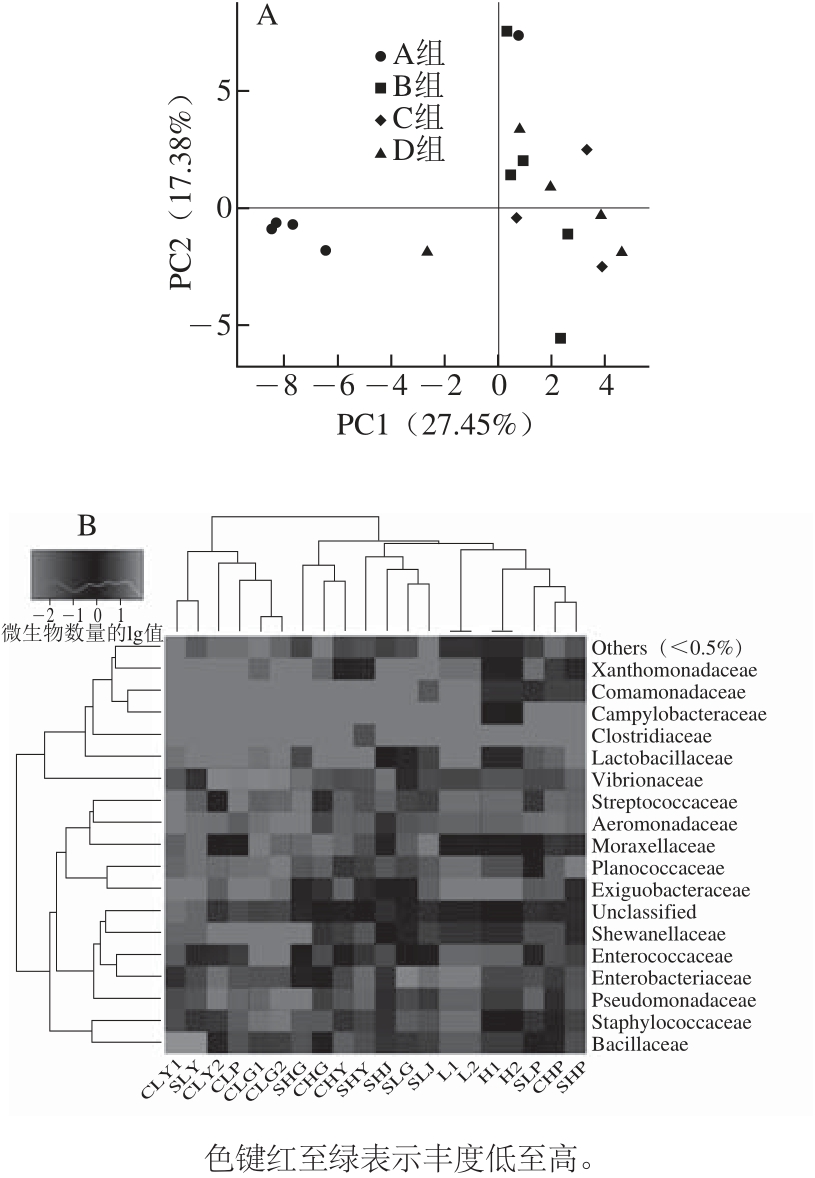
图3 腌干鱼加工过程中细菌OTU的主成分分析图(A)及其科水平分类上的热图(B)B
Fig.3 Principal component analysis (PCA) profile (A) and heatmap at family level (B) of microbial populations in dried-salted fish from different processing stages
由图3可见,4 组样品聚类在一起,PCA获得PC1的贡献率为27.45%,PC2的贡献率为17.38%;从科水平上的丰度热图显示腌干鱼在加工过程中的主要菌群来自黄色单胞菌科(Xanthomondaceae)、Comamonadaceae、弯曲菌科(Campylobacteraceae)、梭菌科(Clostridiaceae)、Lactobacillaceae、Vibrionaceae、Streptococcaceae、Aeromonadaceae、莫拉菌科(Moraxellaceae)、动性球菌科(Planococcaceae)、Exiguobacteraceae、未分类科(Unclassified)、Shewanellaceae、Enterococcaceae、Enterbacteriaceae、Pseudomonadaceae、Staphylococcaceae、Bacillaceae。其中,Staphylococcaceae、Enterbacteriaceae、Aeromonadaceae、Vibrionaceae为优势菌群。
2.3 OUT韦恩图分析
在97%的相似度条件下,得到了每个样品的OTU个数,利用Venn图可以展示多样品共有和各自特有OTU数目,直观展示样品间OTU的重叠情况[24]。结合OTU所代表的物种,可以找出不同环境中的核心微生物。图4a为腌干鱼加工过程细菌OTU venn图分析结果,可以很直观地看出,在不同加工阶段,细菌多样性有很大差异,但各个加工阶段又都含有共同的OUT数,蓝圆鲹在传统腌制加工各阶段中共有4 个OUT,在乳酸菌辅助低盐腌制加工各阶段共有10 个OUT;海鲈在传统腌制加工各阶段中共有23 个OUT,在乳酸菌辅助低盐腌制加工各阶段共有15 个OUT;并且共有的OUT数均占总OUT数的一半以上,说明腌干鱼在不同加工阶段中的核心微生物是一样的。图4b为腌干鱼在两种不同方法加工过程中细菌OTU venn图分析结果,可以很直观的看出,同种鱼在不同加工方法中核心微生物大致相同。
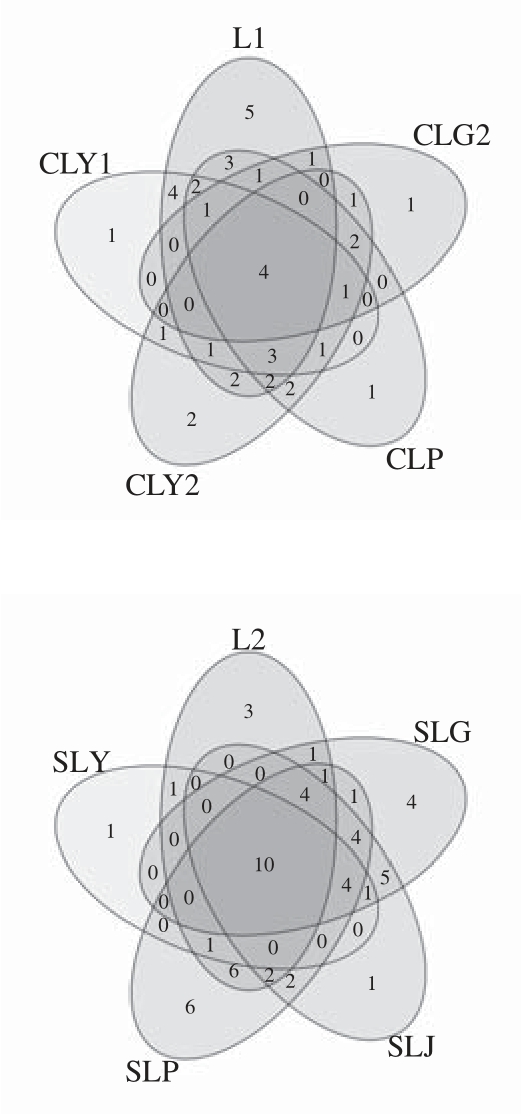
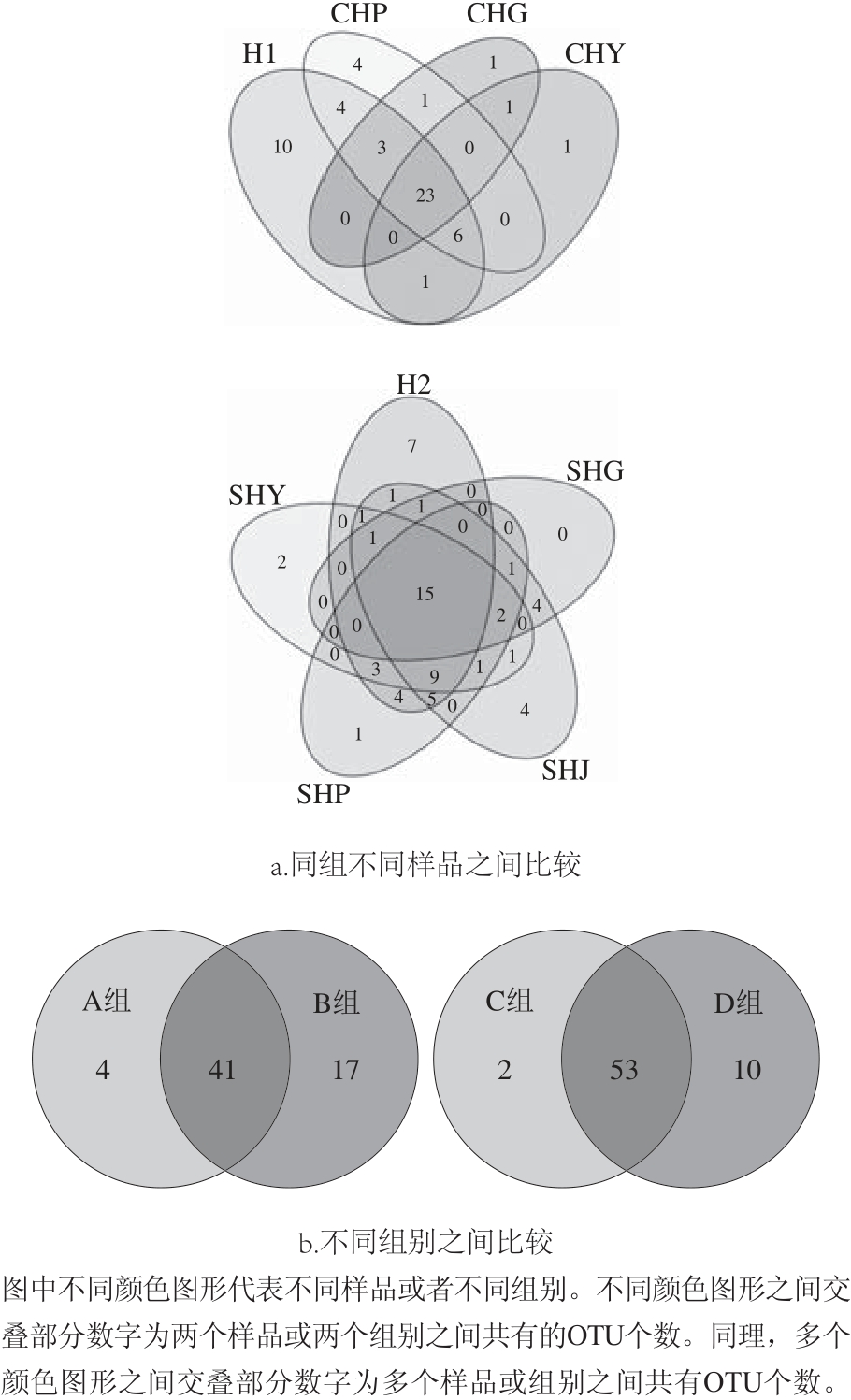
图4 OTU韦恩图分析
Fig.4 Shared OTU across different samples or groups
2.4 微生物群落结构变化
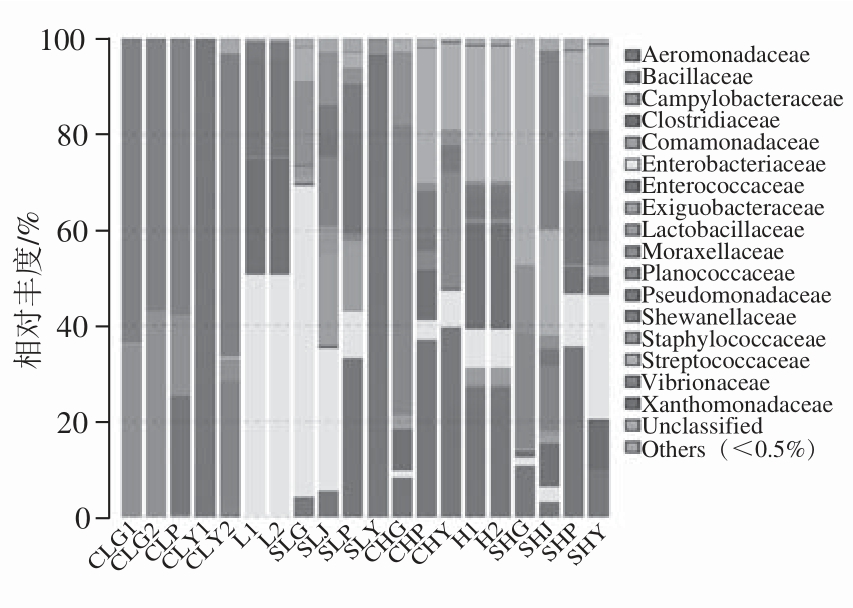
图5 基于科水平腌干鱼加工过程细菌群结构
Fig.5 Family-level community structure of bacteria in dried-salted fish from different processing stages
科水平描述腌干鱼加工过程微生物菌群结构变化如图5所示。本实验自定义相对丰度不小于20%的菌群为优势菌群。由图5蓝圆鰺处理组可知,新鲜蓝圆鲹初始菌相中优势细菌为Enterbacteriaceae、Enterococcactae、Pseudomonadaceae、Shewanellaceae,传统腌制加工过程中菌相比较单一,优势菌主要是Vibrionaceae、Staphylococcaceae、Pseudomonadaceae、Planococcaceae,而乳酸菌辅助腌制加工过程中细菌多样性明显增加,接种乳酸菌发酵后,乳酸菌成为了优势菌,并且促进了Exiguobacteraceae、Staphylococcaceae等优势菌的繁殖。而且还出现了Aeromonadaceae、Lactobacillaceae等传统蓝圆鲹腌制加工过程中没有检测到的菌。由图5海鲈处理组明显看出,海鲈在腌制加工过程中细菌多样性明显高于蓝圆鲹,这与图2 α-多样性指数的箱图显示的结果一致。新鲜海鲈中初始菌相中优势菌主要是Aeromonadaceae、Bacillaceae、Staphylococcaceae、Comamonadaceae、Enterbacteriaceae、Enterococcaceae、Moraganellaceae、Streptococcaceae等,乳酸菌辅助腌制加工过程中细菌多样性也略有增加。
蓝圆鲹在传统腌制过程中Enterbacteriaceae、Enterococcaceae发生了明显变化,在腌制后的过程中,这两种微生物相对丰度减为0,说明腌制过程由于高浓度食盐的渗入可以有效降低腐败菌的繁殖,提高食品的安全品质。在腌制阶段产生了弧菌,并在之后的漂洗和干燥阶段抑制处于优势地位,Vibrionaceae与生物胺的形成息息相关。而接种乳酸菌发酵后,Enterbacteriaceae、Pseudomonadaceae数量有所减少,Vibrionaceae在整个加工过程中也没有检测到,说明接种的乳酸菌发酵剂可以抑制产生物胺菌的生长繁殖,从而降低产品中生物胺含量。乳酸菌和葡萄球菌一跃成为优势菌,研究表明,乳酸菌和葡萄球菌可以影响腌干鱼制品风味的形成。其中葡萄球菌和微球菌能够代谢产生硝酸盐还原酶和过氧化氢酶,形成并稳定色泽[25]。同时,还可以分解蛋白质和脂肪,使得游离氨基酸和脂肪酸以及醛、酮等小分子物质增加,促进产品风味形成。葡萄球菌科的微生物还参与脂肪的降解过程,对脂肪酸的释放有一定的影响。大量的脂肪酸被释放出来,可进一步转化形成甲基酮和醛等挥发性风味成分。乳酸菌不仅含有一般微生物所产生的有关酶,还可以产生 能够分解脂肪酸、亚硝胺,控制内毒素等特殊酶系[26],进而改善产品风味。
海鲈在传统腌制阶段E n t e r o c o c c a c e a e、Pseudomonadaceae数量明显减少,Shewanellaceae、Streptococcaceae、Campylobacteraceae增加,漂洗阶段变化最明显的是Aeromonadaceae增加以及Shewanellaceae相对丰度减少。而干燥阶段的优势菌则为Campylobacteraceae、Planococcaceae、Staphylococcaceae、Streptococcaceae。而接种乳酸菌发酵剂后,海鲈中微生物种类和相对丰度均明显增加,但到干燥阶段随着环境温度的升高及水分活度的减少,其菌相变得较单一,干燥成品中的优势菌仅为Campylobacteraceae、Staphylococcaceae和Streptococcaceae。
综上所述,微生物群落结构不仅与腌制工艺有关,还与鱼的品种有着密切关系。
对腌干鱼加工过程各阶段样品通过试剂盒提取DNA,特异性引物扩增PCR产物,是各个时期各种微生物的16S rDNA的混合物,代表了该加工阶段样品的微生物群体。
由微生物数据库比对分析结果,腌干鱼加工过程反映了细菌种群的多样性特点。其中,蓝圆鲹在初始阶段漫游球菌(Vagococcus)和耶尔森菌(Yersinia)是文库中的优势类别,相对丰度分别为18.75%~40.00%和40.00%~88.23%,随着传统腌制加工的进行,弧菌(Vibrio)成为丰度最高的属类,所占比例为40.00%~97.24%,葡萄球菌(Staphylococcus)成为次要优势菌,所占比例为0%~40.00%。但随着乳酸菌发酵剂的加入,样品中的微生物群落结构发生了变化,弧菌(Vibrio)不再是优势菌,甚至已经消失。未分类的菌属急剧增加,成为主要优势菌,片球菌(Pediococcus)和希瓦氏菌(Shewanella)成为次要优势菌。海鲈在初始阶段菌相比蓝圆鲹复杂,包含11个属的微生物,乳球菌(Lactococcus)、假单胞菌(Pseudomonas)、漫游球菌(Vagocuccus)、耶尔森菌(Yersinia)和未分类的属是文库中的优势菌,最高相对丰度均大于40.00%。随着传统腌制加工的进行,细菌种类并没有明显增加,但物种变化较大。乳球菌(Lactococcus)属和漫游球菌(Vagocuccus)属在整个加工阶段一直处于优势地位,到了腌制和干燥阶段,乳球菌(Lactococcus)的数量达到较大值(42.31%~59.42%),这一阶段微生物最活跃,各种微生物在酶的作用下迅速分解肌肉中的蛋白质和脂肪[27],生成小分子物质,使腌干鱼风味品质变好。期间pH值的降低和水分活度的降低,即使在初始菌相中存在下来的肠杆菌、肠球菌和假单胞菌等活动也会受到抑制,只有抗酸性比较强的细菌才能存活下来。加入乳酸菌辅助发酵后,葡萄球菌属和乳球菌属的相对丰度明显增加,它们在之后的加工过程中将会对制品风味产生良好影响[28]。
腌干鱼在加工过程中微生物群落分析目前还没有前人做过详细分析,但是与生物胺相关的微生物有前人做过相关研究,假单胞菌、弧菌是产生物胺优势微生物,而腌制食品中的生物胺含量较高[29],这为产胺微生物的生长 提供了充足的条件,这可能是假单胞菌和弧菌等是优势菌的重要原因。
蓝圆鲹是海产鱼中的青皮红肉鱼类,肌肉中含血红蛋白较多,因此组氨酸含量也较高,这为产组胺微生物的生长提供了充足的营养物质,所以,组胺生成菌弧菌(Vibrio)和葡萄球菌(Staphylococcus)为蓝圆鲹加工中的优势菌。海鲈 是养殖品种中的白色鱼肉,组氨酸含量明显低于青皮红肉鱼,因此产组胺微生物(弧菌和葡萄球菌)并不占优势。
研究表明,Miseq测序比一般的传统培养方法反映的细菌种类多,能够更好地反映腌制食品体系中细菌菌落结构的多样性及变化,主要是由于MiSeq测序读长和质量可以通过比较每组配对的阅读框的末端片段并拼接成为单个片段的方式得到提高,这使得研究人员使用Illumina MiSeq得到和通过454焦磷酸测序得到的序列长度相似的合并序列,这样不但可以降低测序成本,还可以提高测序结果的质量[30]。
通过宏基因组技术对传统腌干鱼和乳酸 菌法腌干鱼各加工阶段的微生物群落多样性分析表明,加工过程中细菌多样性十分丰富,主要分为三大类:Bacteroidetes、Firmicutes、Proteobacteria;蓝圆鲹初始菌相中优势细菌为Enterbacteriaceae、Enterococcactae、Pseudomonadaceae、Shewanellaceae,新鲜海鲈中初始菌相中优势菌主要是Aeromonadaceae、Bacillaceae、Staphylococcaceae、Comamonadaceae、Enterbacteriaceae、Enterococcaceae、Moraganellaceae、Streptococcaceae等。传统腌制加工过程中菌相比较单一,优势菌主要是Vibrionaceae、Staphylococcaceae、Pseudomonadaceae、Planococcaceae;而乳酸菌法腌制加工过程中 细菌多样性在总体上呈现增加趋势,接种乳酸菌发酵后,乳酸菌成为了优势菌,并且促进了Exiguobacteracea、Staphylococcaceae等优势菌的繁殖。海鲈在腌制加工过程中细菌多样性明显高于蓝圆鲹,乳酸菌法腌制海鲈过程细菌多样性比传统法多,而乳酸菌法腌制蓝圆鲹过程中出现了在传统腌制过程没有的Aeromonadaceae、Lactobacillaceae等菌。
腌干鱼加工过程中细菌多样性与加工方法和鱼的品种有密切关系。乳酸菌法腌干鱼的技术是本实验室开发的快速鱼类腌制加工技术[5-6],目的是在保持腌干鱼制品保持传统腌干鱼特有风味的同时,缩短加工时间,提升产品安全性和风味特性,通过对其微生物多样性分析也进一步证明该技术在加工过程细菌多样性更丰富,特别是有益的微生物成为优势菌,这为乳酸菌法腌干鱼加工技术的进一步优化和推广应用提供技术支持。
参考文献:
[1] 曾令彬, 熊善柏, 王莉. 腊鱼加工过程中微生物及理化特性的变化[J]. 食品科学, 2009, 30(3): 54-57. DOI:10.3321/ j.issn:1002-6630.2009.03.011.
[2] 田国军, 尚艳艳, 黄泽元. 腊鱼中优势乳酸菌的分离、纯化及性质鉴定[J]. 食品与发酵工业, 2011, 37(6): 78-81. DOI:10.13995/ j.cnki.11-180 2/ts.2011.06.033.
[3] 杨锡洪, 吴海燕, 解万翠, 等. 风味咸鱼中乳酸菌和葡萄球菌的分离与鉴定[J]. 食品科学, 2009, 30(21): 192-194. DOI:10.3321/ j.issn:1002-6630.2009.21.045.
[4] 游刚, 吴燕燕, 李来好. 等. 分离自传统腌制鱼类的乳酸菌株发酵特性研究[J]. 食品工业科技, 2014, 35(10): 220-223. DOI:10.13386/ j.issn1002-0306.2014.10.041.
[5] KOPERMSUB P, YUNCHALARD S. Identification of lactic acid bacteria associated with the production of plaa-som, a traditional fermented fi sh product of Thailand[J]. International Journal of Food Microbiology, 2009, 138(3): 200-204.
[6] 吴燕燕, 游刚, 李来好, 等. 低盐乳酸菌法与传统法腌干鱼制品的风味比较[J]. 水产学报, 2014, 38(4): 600-611.
[7] 游刚, 吴燕燕, 李来好, 等. 添加复合乳酸菌再发酵对腌干鱼肉微生物、亚硝酸盐和亚硝胺的影响[J]. 南方水产科学, 2015, 11(4): 109-115. DOI:10.3969/j.issn.2095-0780.2015.04.016.
[8] 王乃富, 李春阳 , 阎征, 等. 乳酸菌发酵对鳙鱼肉糜菌相与品质的影响[J]. 食品科学, 2011, 32(7): 92-96.
[9] SAITHONG P, PANTHAVEE W, BOONYARATANAKORNKIT M, et al. Use of a starter culture of lactic acid bacteria in plaa-som, a Thai fermented fish[J]. Journal of Bioscience and Bioengineering, 2010, 110(5): 553-557. DOI:10.1016/j.jbiosc.2010.06.004.
[10] JO H. Metagenomics: application of genomics to uncultured microorganisms[J]. Microbiology and Molecular Biology Reviews, 2004, 68(4): 669-685. DOI:10.1128/MMBR.68.4.669-685.2004.
[11] 张家松, 段亚飞, 张真真, 等. 对虾肠道微生物菌群的研究进展[J]. 南方水产科学, 2015, 11(6): 114-119. DOI:10.3969/ j.issn.2095-0780.2015.06.016.
[12] HANDELSMAN J, RONDON M R, BRADY S F, et al. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products[J]. Chemistry & Biology, 1998, 5(10): 245-249.
[13] 李建柱, 侯杰, 张鹏飞, 等. 鱼菜共生模式中不同鱼类肠道微生物群落结构的比较[J]. 南方水产科学, 2016, 12(6): 42-50. DOI:10.3969/ j.issn.2095-0780.2016.06.006.
[14] 张岩, 吴燕燕, 李来好, 等. 16S rDNA序列分析鉴定一株合浦珠母贝共附生乳酸菌[J]. 南方水产科学, 2012, 8(6): 9-15. DOI:10.3969/ j.issn.2095-0780.2012.06.002.
[15] POŁKA J, REBECCHI A, PISACANE V, et al. Bacterial diversity in typical Italian salami at different ripening stages as revealed by highthroughput sequencing of 16S rRNA amplicons[J]. Food Microbiology, 2015, 46: 342-356. DOI:10.1016/j.fm.2014.08.023.
[16] WILLIAMS S T, FOSTER P G, LITTLEWOOD D T J. The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny[J]. Gene, 2014, 533(1): 38-47. DOI:10.1016/ j.gene.2013.10.005.
[17] 聂志强, 韩玥, 郑宇, 等. 宏基因组学技术分析传统食醋发酵过程微生物多样性[J]. 食品科学, 2013, 34(15): 198-203. DOI:10.7506/ spkx1002-6630-201315041.
[18] HUSE S M, LES D, HUBER J A, et al. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing[J]. PLoS Genetics, 2008, 4(11): e1000255. DOI:10.1371/journal.pgen.1000255.
[19] TANJA M, L S S. FLASH: fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics, 2011, 27(21): 2957-2963. DOI:10.1093/bioinformatics/btr507.
[20] BOKULICH N A, SATHISH S, FAITH J J, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing[J]. Nature Methods, 2012, 10(1): 57-59. DOI:10.1038/ nmeth.2276.
[21] EDGAR R C, HAAS B J, CLEMENTE J C, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200. DOI:10.1093/bioinformatics/btr381.
[22] 曾燕, 简平, 倪学勤, 等. Illumina MiSeq测序平台测定蒙古羊瘤胃液相和固相菌群多样性[J]. 动物营养学报, 2015, 27(10): 3256-3262. DOI:10.3969/j.issn.1006-267x.2015.10.034.
[23] FADROSH D W, BING M, PAWEL G, et al. An improved dualindexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform[J]. Microbiome, 2014, 2(1): 1-6.
[24] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10(10): 996-998. DOI:10.1038/nmeth.2604.
[25] 吴海燕, 杨磊, 李思东, 等. 生物催化风味在水产品加工中的应用[J]. 广东化工, 2010, 37(7): 66-67. DOI:10.3969/ j.issn.1007-1865.2010.07.030.
[26] 胡彦新, 李清, 王英, 等. 传统发酵食品中产细菌素乳酸菌的筛选与鉴定[J]. 江苏农业科学, 2016, 44(1): 276-278. DOI:10.15889/ j.issn.1002-1302.2016.01.082.
[27] 张会丽, 余翔, 张弘, 等. 鲈鱼风干成熟工艺及对蛋白质水解和感官品质影响[J]. 食品科学, 2010, 31(16): 47-51.
[28] 马欢欢, 吕欣然, 白凤翎, 等. 传统中韩泡菜乳酸菌菌相分析与风味物质组成的比较[J]. 食品与发酵工业, 2015, 41(12): 184-190. DOI:10.13995/j.cnki.11-1802/ts.201512036.
[ 29] 吴燕燕, 陈玉峰. 腌制水产品中生物胺的形成及控制技术研究进展[J]. 食品工业科技, 2014, 35(14): 396-400. DOI:10.13386/ j.issn1002-0306.2014.14.078.
[30] QUAIL M A, IWANKA K, FRANCES S, et al. A large genome center’s improvements to the Illumina sequencing system[J]. Nature Methods, 2008, 5(12): 1005-1010. DOI:10.1038/nmeth.1270.
Microbial Community Diversity in Dried-Salted Fish during Processing Revealed by Illumina MiSeq Sequencing
WU Yanyan1, QIAN Xixi1,2, LI Laihao1, CHEN Shengjun1, DENG Jianchao1, LI Chunsheng1
(1. Key Laboratory of Aquatic Product Processing, Ministry of Agriculture, South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou 510300, China; 2. College of Food Science and Technology, Shanghai Ocean University, Shanghai 201306, China)
Abstract:In this study, we investigated the microbial variations during the processing of traditional salted fish and lactic acidfermented fish, aiming to provide a theoretical basis for the new processing technology. Illumina MiSeq sequencing was applied to analyze the microbial community diversity in dried-salted fish during different processing stages. The results indicated that the processing system contained highly diverse bacteria, which could be mainly divided into three categories: Bacteroidetes, Firmicutes and Proteobacteria. Enterbacteriaceae, Enterococcactae, Pseudomonadacea and Shewanellaceae were dominant microorganisms in brown-striped mackerel scad (Decapterus maruadsi) during the initial stages of processing while Aeromonadaceae, Bacillaceae, Staphylococcaceae, Comamonadaceae, Enterbacteriaceae, Enterococcaceae, Moraganellaceae and Streptococcaceae were dominant microorganisms in sea bass (Lateolabrax japonicus). The microbial community was homogeneous during the traditional curing process, and the dominant microorganisms identified were Vibrionaceae, Staphylococcaceae, Pseudomonadaceae and Planococcaceae. After inoculation of lactic acid bacteria, the microbial diversity during the fermentation process showed an increasing trend, and lactic acid bacteria became dominant and promoted the growth of the dominant bacteria Exiguobacteracea and Staphylococcaceae; Aeromonadaceae and Lactobacillaceae, which did not appear during the traditional processing, were detected. Microbial community diversity in sea bass during the curing process was significantly higher than thatin brown-striped mackerel scad. Microbial community diversity was also increased markedly during the processing of lactic acidfermented fish.
Key words:dried-salted fish; Illumina MiSeq sequencing; microbial community diversity; dominant bacteria
DOI:10.7506/spkx1002-6630-201712001
收稿日期:2016-06-12
基金项目:国家自然科学基金面上项目(31371800;31571869);广东省海洋渔业科技推广专项(A201301C01);广东省海洋渔业科技与产业发展专项(A201501C02)
作者简介:吴燕燕(1969—),女,研究员,博士,主要从事水产品加工与质量安全研究。E-mail:wuyygd@163.com
中图分类号:TS254.1
文献标志码:A
文章编号:1002-6630(2017)12-0001-08
引文格式:吴燕燕, 钱茜茜, 李来好, 等. 基于Illumina MiSeq技术分析腌干鱼加工过程中微生物群落多样性[J]. 食品科学, 2017, 38(12): 1-8.
DOI:10.7506/spkx1002-6630-201712001. http://www.spkx.net.cn WU Yanyan, QIAN Xixi, LI Laihao, et al. Microbial community diversity in dried-salted fish during processing revealed by Illumina MiSeq sequencing[J]. Food Science, 2017, 38(12): 1-8. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201712001. http://www.spkx.net.cn