同源重组法构建副干酪乳杆菌组氨酸蛋白激酶基因缺失突变株
岳元春1,2,王 洋1,2,由 田1,2,高冬妮1,2,平文祥1,2,葛菁萍1,2,*
(1.黑龙江大学生命科学学院,微生物省高校重点实验室,黑龙江 哈尔滨 150080;2.黑龙江大学 农业微生物技术教育部工程研究中心,黑龙江 哈尔滨 150500)
摘 要:构建副干酪乳杆菌(Lactobacillus paracasei)组氨酸蛋白激酶prcK基因缺失突变株,为prcK基因功能研究提供实验工具。采用同源重组技术构建质粒pKLKRT(含prcK:Tetr基因),将其电转化进入L. paracasei HD1.7中,使prcK:Tetr基因同源重组到L. paracasei HD1.7的染色体上,通过四环素耐药、氨苄青霉素敏感等特性筛选出含有prcK:Tetr基因的新L. paracasei HD1.7。结果表明,经聚合酶链式反应(polymerase chain reaction,PCR)验证及酶切验证后确定质粒pKLKRT构建成功,并成功转化入L. paracasei HD1.7中,经PCR确认L. paracasei HD1.7 prcK基因缺失突变株构建成功。该突变株产生的细菌素效价比出发菌株低23.61%。采用同源重组方法成功构建L. paracasei HD1.7 prcK基因缺失突变株,为研究L. paracasei HD1.7群体效应相关基因的分子机理奠定基础。
关键词:副干酪乳杆菌;细菌素;组氨酸蛋白激酶基因;同源重组
乳酸菌常常与发酵食品及饮品联系在一起[1-3]。本实验所研究的副干酪乳杆菌(Lactobacillus paracasei)HD1.7是从乳酸酸菜发酵液中分离出的一株乳酸菌,经研究发现,该菌株所产细菌素可以有效抑制多种革兰氏阳性菌、革兰氏阴性菌和酿酒酵母[4-6],而该细菌素的产生受一种被称为三组分调节系统(three-component regulatory system,3CRS)的体系调控[7-9]。系统由3 个共转录基因组成,分别为被分泌至细胞外的细菌素类似物自诱导肽(autoinducer protein,AIP)和一个标准的细菌双组分调节系统(two-component regulatory system,2CRS),系统由一个组氨酸蛋白激酶(histidine protein kinase,HPK)和一个反应调节因子(response regulator,RR)组成[10-11],其中2CRS是细菌群体效应现象(quorum sense,QS)的调控系统。由田[12]从L. paracasei HD1.7中克隆到组氨酸蛋白激酶基因prcK,其无论在结构还是功能上均与已探明功能的低GC含量革兰氏阳性菌QS系统中的HPK家族中HPK10亚家族十分相似,相似性为99%,其蛋白产物为L. paracasei HD1.7群体效应系统中的HPK,负责调控细菌素的生成。
同源重组是任何一段具有同源序列的2个基因DNA之间的交换,涉及到参与重组的双方染色体或DNA分子的断裂和重接,在基因工程中已有广泛的应用[13-20]。本研究利用同源重组的方法敲除prcK基因,试图摸索一套完善的L. paracasei prcK基因缺失菌株的构建方法,为研究L. paracasei群体效应相关基因的功能及分子机理奠定基础,对于提高L. paracasei的应用领域及开展L. paracasei蛋白组研究也具有非常重要的意义。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
L. paracasei HD1.7、大肠杆菌(Escherichia coli)DH5α、枯草芽孢杆菌(Bacillus subtilis),以及质粒pUC18(2 686 bp,Ampr)均由黑龙江大学微生物重点实验室提供;质粒pUC18-tet(4 086 bp,Tetr)由本实验室自行构建;克隆载体pMD18-T(2 692 bp,Ampr)购自大连宝生物工程有限公司。
1.1.2 培养基
MRS液体培养基:大豆蛋白胨10 g、牛肉膏10 g、酵母提取物5 g、D-葡萄糖20 g、磷酸氢二钾2 g、亚硫酸钠0.1 g、乙酸钠5 g、硫酸镁0.2 g、硫酸锰0.05 g、柠檬酸铵2 g,溶于800 mL ddH2O中,加入吐温-801 mL,用浓硫酸调节pH 5.5,补足水至1 000 mL,115 ℃高压灭菌15 min。
MRS固体培养基:1 000 mL MRS液体培养基中加入琼脂粉15~20 g,121 ℃高压灭菌15 min。
LB液体培养基:胰蛋白胨10 g,酵母提取物5 g,NaCl10 g,溶于800 mL ddH2O中,用10 mol/mL NaOH调pH值至7.0,补足水至1 000 mL,121 ℃高压灭菌15 min。
LB固体培养基:1 000 mL LB液体培养基中加入琼脂粉12~15 g,121 ℃高压灭菌15 min。
1.1.3 试剂
Taq DNA聚合酶、PrimeSTAR®HS DNA聚合酶、T4 DNA连接酶、RNase A(DNase free)、限制性内切酶、抗生素(氨苄青霉素、四环素) 大连宝生物工程有限公司;细菌基因组DNA(小量)抽提试剂盒、胶回收(小量)试剂盒、质粒(小量)抽提试剂盒 上海华舜生物技术有限公司。
1.1.4 引物
引物由大连宝生物工程有限公司合成(表1)。
表1 引物的设计
Table1 Primer sequences used in this study

注:下划线表示酶切位点。
CCGGAGCTCTACCTTAATGATTTAGATGCGAGCGSacⅠ用于构建自杀质粒pKLKRT的一侧同源臂(1.34 kb)prcKL-down引物名称引物序列(5’→3’)限制性内切酶用途prcK-upATGGAAACTTATTCTGATCTAGCCT用于获得prcK基因全长(1.3 kb)prcK-downAAGTCATCTCCCTATAAACAAAGTG prcKL-up GTCGGTACCGGTTTTGCCGTCATCAGCGCACTTGKpnⅠ用于构建自杀质粒pKLKRT的一侧同源臂(1.37 kb)prcKR-down GTCGGTACCGATTGTTCCTTCGGTGTGGATGTGTKpnⅠprcKR-up GTCGGTACCTAATAGATATGTTCTGCCAAGGGTKpnⅠCCGGGTACCTCTCATGTTTGACAGCTTKpnⅠ用于获得四环素抗性基因全长(1.4 kb)Tet-down CCGCTGCAGACTAATCAGCTGGACTAAGGTGTATPstⅠTet-up
1.2 仪器与设备
Air TECH超静工作台 苏净集团安泰公司;ABI9700聚合酶链式反应(polymerase chain reaction,PCR)仪 基因有限公司;Omega10凝胶成像分析系统美国Ultralum公司;TGL-16G高速台式离心机 上海安亭科学仪器厂;HH-B11-420-S电热恒温培养箱 上海跃进医疗器械厂;DYY-Ⅲ-8B电泳仪 北京六一仪器厂;FE20 pH计 梅特勒-托利多公司;Gene Pulser X-cell电穿孔仪 美国Bio-Rad公司。
1.3 方法
1.3.1 prcK基因的克隆
参照GenBank中的L. paracasei E93490 prcK基因序列,利用软件Primer 5.0设计一对引物:prcK-up/ prcK-down,模板L. paracasei HD1.7基因组用细菌基因组DNA抽提试剂盒提取,PCR体系(50 μL):5×PrimeSTAR Buffer(Mg2+plus)10 μL、dNTP Mixture(2.5 mmol/L)4 μL、prcK-up(1 pmol/μL)10 μL、prcK-down(1 pmol/μL)10 μL、模板(25 ng/μL)10 μL、PrimeSTAR HS DNA聚合酶(2.5 U/μL)0.5 μL、ddH2O 5.5 μL。PCR扩增条件:98 ℃预变性3 min;98 ℃变性10 s,54 ℃退火15 s,72 ℃延伸1 min 30 s,30 个循环;72 ℃延伸10 min。
1.3.2 prcK基因同源臂的克隆
根据prcK全基因序列设计两对引物prcKL-up/prcKL-down和prcKR-up/prcKR-down,在prcKL-up和prcKL-down分别引入限制性酶切位点SacⅠ和KpnⅠ,prcKR-up和prcKR-down分别引入限制性酶切位点KpnⅠ和PstⅠ。以L. paracasei HD1.7基因组DNA为模板,使用上述2 对引物PCR扩增prcK基因的上游(L端)序列和下游(R端)序列。PCR体系同1.3.1节。PCR扩增条件:98 ℃预变性3 min;98 ℃变性10 s,62 ℃(L端)或55 ℃(R端)退火15 s,72 ℃延伸1 min 30 s,30 个循环;72 ℃延伸10 min。
1.3.3 prcK基因敲除质粒的构建
从E. coli DH5α中提取质粒pUC18,分别用限制性内切酶SacⅠ及KpnⅠ酶切,并在T4连接酶的作用下与1.3.2节中扩增出的prcK基因L端序列(1.34 kb)连接,构建载体pUC18-KL;随后将载体pUC18-KL分别用限制性内切酶KpnⅠ及PstⅠ酶切,并与扩增出的prcK基因R端序列(1.37 kb)连接,构建载体pUC18-KLKR;最后将质粒pUC18-tet(Ampr,Tetr)及载体pUC18-KLKR用KpnⅠ酶切,并将酶切产物Tetr基因片段(1.4 kb)及pUC18-KLKR片段(5.3 kb)连接,获得的prcK基因缺失质粒,命名为pKLKRT。
1.3.4 电转化
将上述5 μL基因缺失质粒(0.3~0.5 μg)与40 μL L. paracasei HD1.7感受态细胞冰浴条件下混匀,然后将其加入到冰预冷的0.2 cm电转杯中,并于冰上放置10 min。使用电穿孔仪,设定各参数:电阻值200 Ω,电容值25 μF,点击时间5 ms,电压1.8 kV。电穿孔后迅速加入500 μL MRS培养基,转入1.5 mL离心管内,30 ℃条件下温育3 h。取100 μL涂布于相应的抗生素筛选平板上,30 ℃培养72 h,使基因缺失质粒与细菌染色体同源重组。
1.3.5 同源重组转化子的检测
根据1.3.1节的方法提取转化子基因组DNA,以转化子基因组DNA为模板,用引物对prcKL-up/prcKR-down和Tet-up/Tet-down检测转化子,同时,用出发菌株基因组作对照。
prcKL-up/prcKR-down引物对PCR体系、扩增条件同1.3.1节;Tet-up/Tet-down引物对PCR体系同1.3.1节,PCR扩增条件为:98 ℃预变性3 min;98 ℃变性10 s,46 ℃退火15 s,72 ℃延伸1 min 30 s,30 个循环;72 ℃延伸10 min。
1.3.6 L. paracasei HD1.7 prcK基因缺失突变株发酵液抑菌效果检测
对突变株和出发菌株L. paracasei HD1.7(对照)进行发酵,做抑菌实验,指示菌选B. subtilis。然后对比突变株和出发菌株发酵上清液对指示菌的抑制程度,具体方法见参考文献[21-23]。
2 结果与分析
2.1 prcK基因的克隆
按1.3.1节所述进行PCR扩增,PCR扩增产物经胶回收试剂盒进行回收,以1%的琼脂糖凝胶电泳检测,可见PCR产物大小与预期大小1.3 kb相符(图略)。将PCR产物连入载体pMD18-T,转化入E. coli DH5α感受态细胞后,进行蓝白斑筛选。重组克隆质粒经PCR鉴定和酶切鉴定正确后进行测序。测序结果与GenBank上公布的序列进行相似性比较,相似性达99%,可以确定L. paracasei HD1.7基因组上存在prcK基因。
2.2 prcK基因同源臂的克隆
按1.3.2节所述扩增prcK靶基因的同源臂L端和R端,PCR扩增产物经胶回收试剂盒进行回收,以1%的琼脂糖凝胶电泳检测,PCR产物L端和R端均与预期大小1.34 kb和1.37 kb相符,如图1、2所示。将L端和R端均连入pMD18-T载体,转化入E. coli DH5α感受态细胞后,进行蓝白斑筛选。L端重组质粒经PCR鉴定和SacⅠ和KpnⅠ的双酶切鉴定后证实克隆成功;R端重组质粒经PCR鉴定和KpnⅠ和PstⅠ的双酶切鉴定后证实克隆成功。

图 11 pprrccKKLL--uupp//pprrccKKL-down引物对PCR结果
Fig.1 Results of PCR amplification with prcKL-up and prcKL-down primers
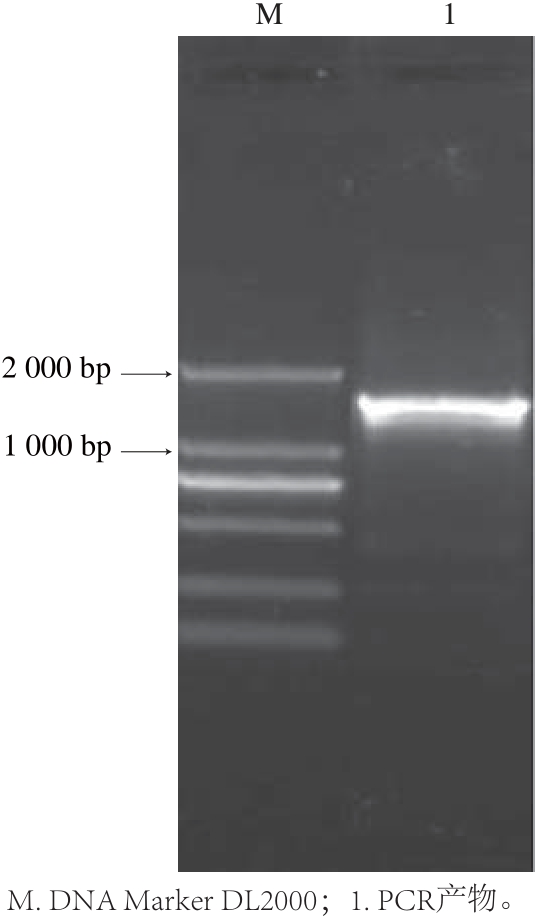
图2 2 prcKprcKR-up/R-up/prcKprcKR-down引物对PCR结果PCR
Fig.2 Results of PCR amplification with prcKR-up and prcKR-down primers
2.3 prcK基因缺失质粒的构建
将质粒pUC18-tet(Ampr、Tetr)及重组质粒pUC18-KLKR用KpnⅠ酶切,并将酶切产物Tetr片段(1.4 kb)及pUC18-KLKR(5.3 kb)线性片段连接,连接产物转化至E. coli DH5α感受态细胞中,四环素、氨苄青霉素抗性平板筛选。
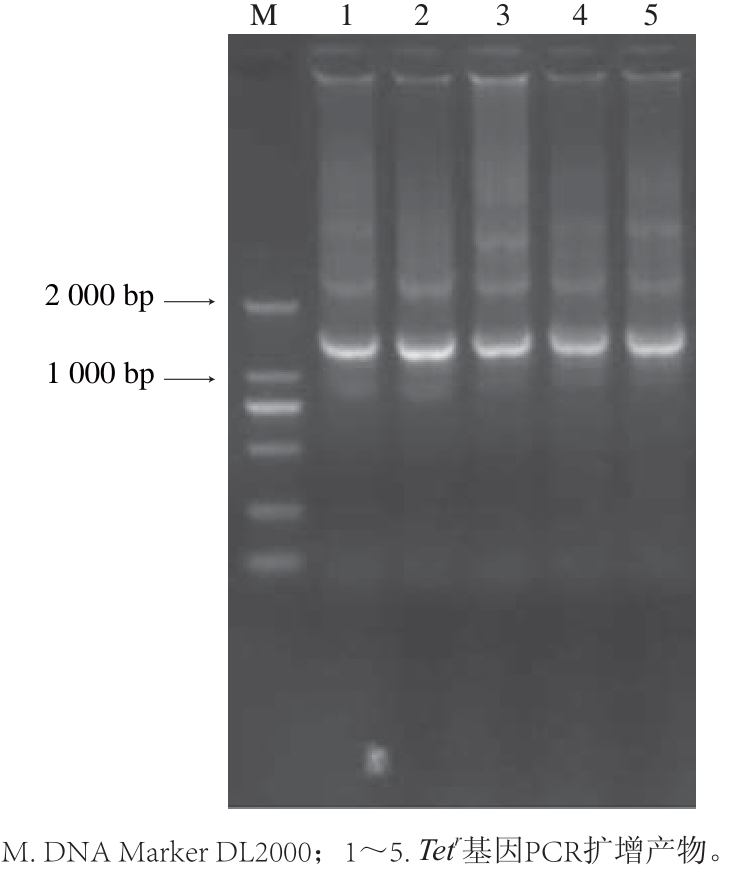
图3 自杀质粒PCR扩增Tetr基因结果
Fig.3 PCR identification of recombination plasmid
对得到的阳性转化子进行PCR验证和酶切验证。PCR验证:以菌液为模板,Tet-up/Tet-down为引物,进行PCR反应,1~5泳道均在1.4 kb处有清晰条带,与Tetr基因大小相符(图3);酶切验证:用限制性内切酶KpnⅠ酶切pKLKRT质粒,在第2、5泳道产生大小约为5.3 kb和 1.4 kb的2 个片段(图4),大小分别与重组质粒pUC18-KLKR和Tetr相符;用限制性内切酶PstⅠ酶切pKLKRT质粒,在第2、5泳道产生大小约为6.7 kb的单一条带(图5)。由图5可知,1、3和4号菌株为假阳性,而2、5号为阳性转化子。证明Tetr基因已经重组到重组质粒pUC18-KLKR上,重组质粒pKLKRT构建正确。
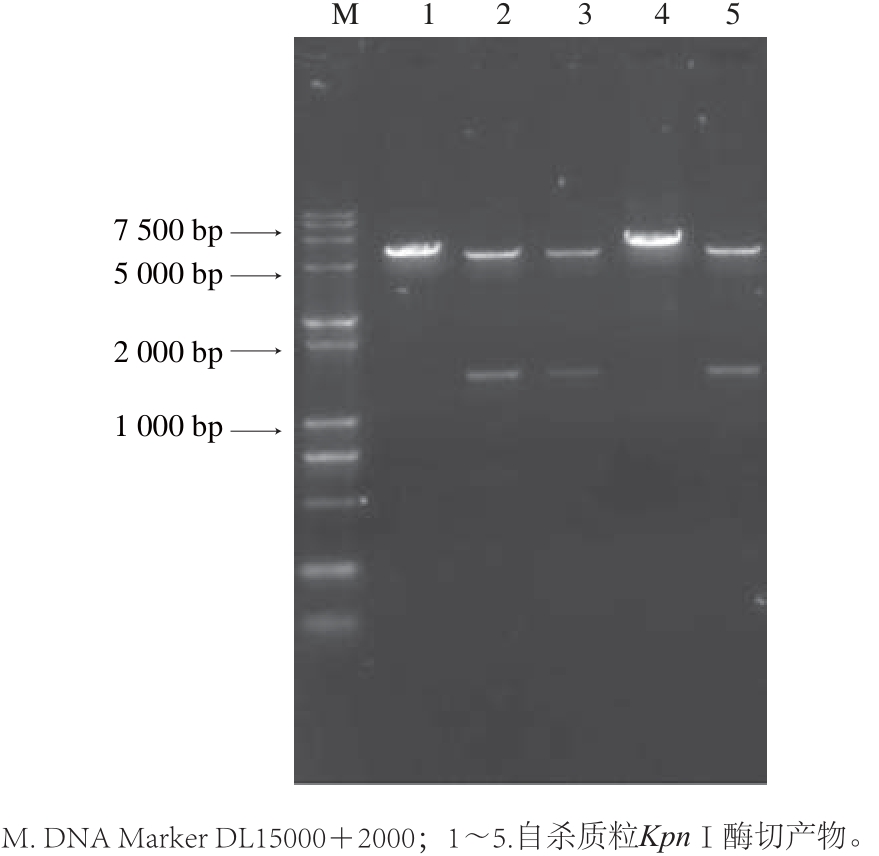
图4 自杀质粒KpnⅠ酶切验证结果
Fig.4 Identification of recombination plasmid with KpnⅠ digestion
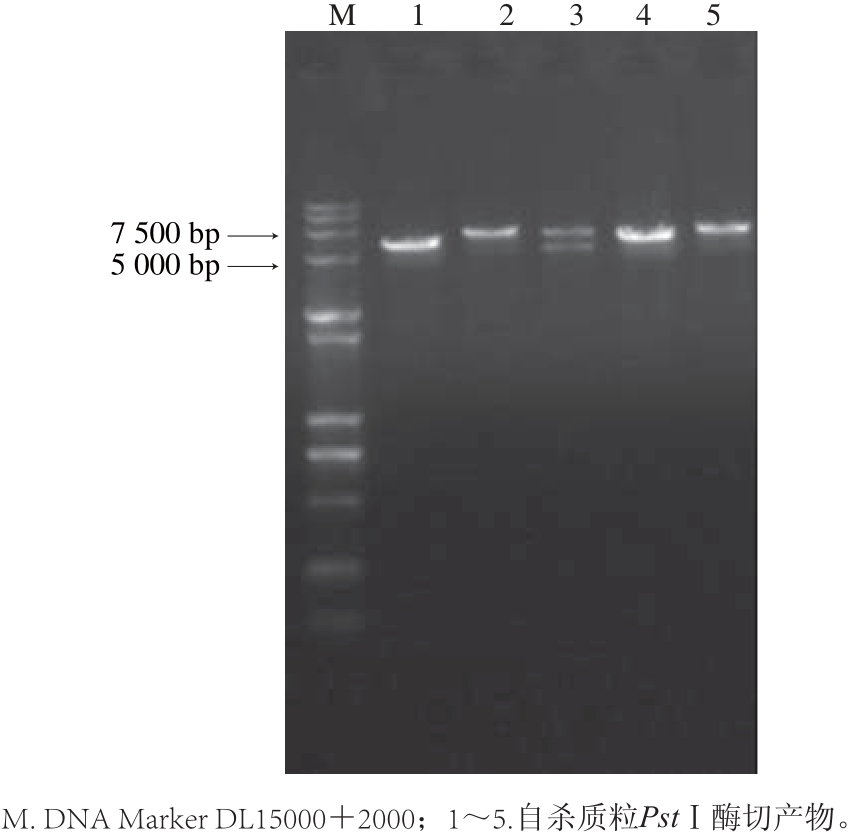
图5 自杀质粒PstⅠ酶切验证结果
Fig.5 Identification of recombination plasmid with PstⅠ digestion
2.4 prcK基因缺失突变株的筛选与鉴定
在抗性平板中挑选出能在四环素(50 µg/mL)平板上生长而不能在氨苄青霉素(100 µg/mL)平板上生长的突变株,它是由导入其细胞内的重组质粒pKLKRT与其基因组DNA发生同源重组而产生的突变株,命名为“ΔK”。

图6 6 TetTet-up/-up/TetTet-down引物对PCR验证结果R
Fig.6 The results of PCR identification with Tet-up and Tet-down primers

图7 7 prcKprcKL-up/L-up/prcKprcKR-down引物对PCR验证结果
Fig.7 PCR identification with prcKL-up and prcKR-down primers
以突变株基因组DNA为模板,出发菌株L. paracasei HD1.7为对照菌株,Tet-up/Tet-down为引物,进行PCR验证,从突变株基因组DNA中扩增出与Tetr基因(1.4 kb)大小相同的片段,而不能从出发菌株中扩增出相同的片段(图6);同理以prcKL-up/prcKR-down为引物,进行PCR,以出发菌株基因组DNA为模板,扩增出的片段约2.7 kb,而以突变株基因组DNA为模板,扩增出的片段为L端、R端和Tetr基因的总和约4.1 kb(图7)。PCR检测证实转化子Δ K为prcK基因缺失突变株。
综上所述,重组质粒pKLKRT与L. paracasei HD1.7基因组成功发生同源重组,即重组质粒的两个同源臂与L. paracasei HD1.7基因组发生双交换,从而将Tetr基因插入基因组中,同时阻断prcK基因,使其无法正确表达目的产物。
2.5 L. paracasei HD1.7 prcK基因缺失突变株发酵液抑菌效果

图8 突变株和出发菌株对枯草芽孢杆菌的抑菌实验
Fig.8 Antibacterial efficacies of the original strain and prcK deletion mutant strain against B. subtilis
对突变株及出发菌株L. paracasei HD1.7(作对照)进行发酵,做抑菌实验,指示菌选用B. subtilis,结果如图8所示,prcK基因缺失突变株发酵液对枯草芽孢杆菌的抑制程度比出发菌株的抑制程度弱,其产生的细菌素效价比出发菌株低23.61%,说明突变株所产生细菌素的量比出发菌株低。
3 讨 论
自杀质粒是进行基因敲除的有效工具,它的构建必须符合一定的条件,如载体进入宿主细胞后不能自我复制,只有通过与宿主基因组发生同源重组,从而整合进入宿主基因组。同源重组现象在生物的进化过程中是较为常见的,但其发生的概率却很低,其发生概率与同源序列长短息息相关[24-27],在一定范围内同源序列的长度越长,发生概率越高,对于细菌而言,两条同源臂长度超过200 bp就有发生同源重组的可能。Leloup等[28]研究了清酒乳杆菌(Lactobacillus sake)中同源片段长度对同源单交换的影响,发现在同源片段长度0.3~1.2 kb范围内同源重组的效率呈对数增长,1.2 kb以后同源重组的效率增高不明显,而且不同位置的DNA同源重组的效率不同。李昌瑜等[29]分别对SP12基因及SP20基因进行敲除,其一侧同源重组片段长度分别为1 kb和600 bp,另一侧同源重组片段长度差异不大,结果发现SP12基因同源重组率为50%,而SP20基因同源重组率为20%,可见同源重组片段任何一侧片段的延长,均可导致同源重组效率的提高。
N a k a y a m a等[30]利用反向P C R技术克隆得到L. paracasei HD1.7拟群体效应相关基因prcK。本研究根据该文献提供的基因序列设计一对引物克隆得到目的基因prcK,以窄宿主范围的质粒pUC18为骨架载体,利用可在G+中表达的四环素抗性基因作为选择标记,构建L. paracasei HD1.7 prcK基因缺失质粒pKLKRT。考虑到同源臂长短对同源重组发生概率的影响,本研究设计的自杀质粒的同源臂是以目的基因及其两翼序列为基础,将标记基因插入2条同源臂中间,2条同源臂分别与其同源区域互换,这种同源臂的设计方式使得同源臂的长短可以较随意的调整,有利于同源重组的发生,且可以自行设计合适的酶切位点供标记基因插入同源臂之间,提高同源重组发生的概率,有效增加基因缺失突变株构建的可能。对L. paracasei HD1.7 prcK基因缺失突变株进行抑菌实验发现,其对B. subtilis的抑菌效果明显小于出发菌株,细菌素效价比出发菌株降低23.61%,说明prcK基因的缺失对L. paracasei HD1.7细菌素的产生有一定的影响,其在L. paracasei HD1.7群体效应现象中扮演重要的角色。
参考文献:
[1] DOMÍNGUEZ-MANZANO J, JIMÉNEZ-DÍAZ R. Suppression of bacteriocin production in mixed-species cultures of lactic acid bacteria[J]. Food Control, 2013, 30(2): 474-479. DOI:10.1016/ j.foodcont.2012.09.014.
[2] DEMERS-MATHIEUA V, ST-GELAISA D, AUDY J, et al. Effect of the low-fat Cheddar cheese manufacturing process on the viability of Bifidobacterium animalis subsp. lactis, Lactobacillus rhamnosus, Lactobacillus paracasei/casei, and Lactobacillus plantarum isolates[J]. Journal of Functional Foods, 2016, 24: 327-337. DOI:10.1016/ j.jff.2016.04.025.
[3] XIN L, LAN H Y, JING D, et al. Purification of novel bacteriocin produced by Lactobacillus coryniformis MXJ 32 for inhibiting bacterial foodborne pathogens including antibiotic-resistant microorganisms[J]. Food Control, 2014, 46: 264-271. DOI:10.1016/j.foodcont.2014.05.028.
[4] WANG S L, HUANG T Y, WANG C Y, et al. Bioconversion of squid pen by Lactobacillus paracasei subsp. paracasei TKU010 for the production of proteases and lettuce growth enhancing biofertilizers[J]. Bioresource Technology, 2008, 99(13): 5436-5443. DOI:10.1016/ j.biortech.2007.11.006.
[5] WANG C Y, LIN P R, NG C C, et al. Probiotic properties of Lactobacillus strains isolated from the feces of breast-fed infants and Taiwanese pickled cabbage[J]. Anaerobe, 2010, 16(6): 578-585. DOI:10.1016/j.anaerobe.2010.10.003.
[6] WANNUN P, PIWAT S, TEANPAISAN R. Purification and characterization of bacteriocin produced by oral Lactobacillus paracasei SD1[J]. Anaerobe, 2014, 27: 17-21. DOI:10.1016/j.anaerobe.2014.03.001.
[7] LI X, HAN Y, YANG Q, et al. Detection of quorum sensing signal molecules and mutation of luxS gene in Vibrio ichthyoenteri[J]. Research in Microbiology, 2010, 161(1): 51-57. DOI:10.1016/j.resmic.2009.10.004.
[8] GE J P, WANG Y, GAO D N, et al. Effect of small interfering RNA against Paracin 1.7 bacteriocin produced by Lactobacillus paracasei HD1-7[J]. Journal of Basic Microbiology, 2015, 55: 1-7. DOI:10.1002/ jobm.201400938.
[9] KLEEREBEZEM M. Quorum sensing control of lantibiotic production; nisin and subtilin autoregulate their own biosynthesis[J]. Peptides, 2004, 25(9): 1405-1414. DOI:10.1016/j.peptides.2003.10.021.
[10] WEST A H, STOCK A M. Histidine kinases and response regulator proteins in two-component signalling systems[J]. Trends in Biochemical Sciences, 2001, 26(6): 369-376. DOI:10.1016/S0968- 0004(01)01852-7.
[11] RUCHI A, PREM K V, HARINI R, et al. FRET reveals multiple interaction states between two component signalling system proteins of M. tuberculosis[J]. Biochimica et Biophysica Acta, 2016, 1860:1498-1507. DOI:10.1016/j.bbagen.2016.04.011.
[12] 由田. Lactobacillus paracasei HD1.7 prcR、prcK基因缺失突变株的构建[D]. 哈尔滨: 黑龙江大学, 2012: 58-73.
[13] LORENZ M C, MUIR R S, LI M E , et al. Gene disruption with PCR products in Saccharomy cescerevisiae[J]. Gene, 1995, 158 (1): 113-117.
[14] GULEDNER U, HECK S, PLEDER T, et al. A new efficient gene disruption cassette for repeated use in budding yeast[J]. Nucleic Acids Research, 1996, 24(13): 2519-2524. DOI:10.1093/nar/24.13.2519.
[15] LAU P C Y, SUNG C K, LEE J H, et al. PCR ligation mutagenesis in transform able streptococci: application and efficiency[J]. Journal of Microbiological Methods, 2002, 49(2): 193-205. DOI:10.1016/S0167-7012(01)00369-4.
[16] MINÉ-HATTAB J, ROTHSTEIN R. Gene targeting and homologous recombination in Saccharomyces cerevisiae[M]//Topics in Current Genetics. Netherlands: Springer, 2013: 71-89. DOI:10.1007/978-94-007-4531-5_3.
[17] BOSE J L, FEY P D, BAYLES K W. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus[J]. Applied and Environmental Microbiology, 2013, 79(7): 2218-2224. DOI:10.1128/ AEM.00136-13.
[18] ISHIZAKI K, JOHZUKA-HISATOMI Y, ISHIDA S, et al. Homologous recombination-mediated gene targeting in the liverwort Marchantia polymorpha L.[J]. Scientific Reports, 2013, 3(6): 1-6. DOI:10.1038/ srep01532.
[19] VEMULAWADA C, SWAGAT K P, RUDRA P P, et al. Establishing targeted carp TLR22 gene disruption via homologous recombination using CRISPR/Cas9[J]. Development and Comparative Immunology, 2016, 61: 242-247. DOI:10.1016/j.dci.2016.04.009.
[20] ZELENSKY A N, SANCHEZ H, RISTIC D, et al. Caffeine suppresses homologous recombination through interference with RAD51-mediated joint molecule formation[J]. Nucleic Acids Research, 2013, 41(15): 1-15. DOI:10.1093/nar/gkt375.
[21] 房保柱. 副干酪乳杆菌HD1.7的群体感应现象研究及与拮抗菌的生态学关系[D]. 哈尔滨: 黑龙江大学, 2012: 21-22.
[22] GE J P, PING W X, SONG G, et al. Paracin 1.7, a bacteriocin produced by Lactobacillus paracasei HD1.7 isolated from Chinese cabbage sauerkraut, a traditional Chinese fermented vegetable food[J]. Acta Microbiologica Sinica, 2009, 49(5): 609-616. DOI:10.13343/j.cnki. wsxb.2009.05.009.
[23] 葛菁萍, 房保住, 苑婷婷, 等. 副干酪乳杆菌HD1.7群体感应行为[J]. 微生物学报, 2011, 51(11): 1561-1567. DOI:10.13343/j.cnki.wsxb.2011.11.017.
[24] AKHUNOV E D, GOODYEAR A W, GENG S, et al. The organization and rate of evolution of wheat genomes are correlated with recombination rates along chromosome arms[J]. Genome Research, 2003, 13(5): 753-763. DOI:10.1101/gr.808603.
[25] AKHUNOV E D, AKHUNOVA A R, LINKIEWICZ A M, et al. Synteny perturbations between wheat homoeologous chromosomes caused by locus duplications and deletions correlate with recombination rate[J]. PNAS, 2003, 100(19): 10836-10841. DOI:10.1073/pnas.1934431100.
[26] LEI Z W, ARTHUR S L, LI L, et al. Transcription-coupled homologous recombination after oxidative damage[J]. DNA Repair, 2016, 44: 76-80. DOI:10.1016/j.dnarep.2016.05.009.
[27] HOFFA G, BERTRANDA C, PIOTROWSKI E, et al. Implication of RuvABC and RecG in homologous recombination in Streptomyces ambofaciens[J]. Research in Microbiology, 2016, 168(1): 26-35. DOI:10.1016/j.resmic.2016.07.003.
[28] LELOUP L, EHRLICH S D, ZAGOREC M, et al. Single-crossover integration in the Lactobacillus sake chromosome and insertional inactivation of the ptsI and lacL genes[J]. Applied and Environmental Microbiology, 1997, 63(6): 2117-2123.
[29] 李昌瑜, 胡赛阳, 王行国, 等. 同源重组法敲除酵母snoRNA基因的初步探讨[J]. 湖北大学学报(自然科学版), 2007, 29(4): 396-400.
[30] NAKAYAMA J, AKKERMANS A D L, de VOS W M. High-throughput PCR screening of genes for three-component regulatory system putatively involved in quorum-sensing from low G+C Gram-positive bacteria[J]. Bioscience Biotechnology & Biochemistry, 2003, 67(3): 480-489. DOI:10.1271/bbb.67.480.
Construction of prcK Gene Deletion Mutant of Lactobacillus paracasei by Homologous Recombination
YUE Yuanchun1,2, WANG Yang1,2, YOU Tian1,2, GAO Dongni1,2, PING Wenxiang1,2, GE Jingping1,2,*
(1. Key Laboratory of Microbiology, Life Science College, Heilongjiang University, Harbin 150080, China; 2. Engineering Research Center of Agricultural Microbiology Technology, Ministry of Education, Heilongjiang University, Harbin 150500, China)
Abstract:This study aimed to construct a histidine protein kinase gene (prcK) deletion mutant of Lactobacillus paracasei HD1.7 for providing an experiment tool for research on the function of the prcK gene. In this research, homologous recombination method was used. Plasmid pKLKRT (including prcK:Tetr) was constructed and used to transform L. paracasei HD1.7 by eletroporation. The prcK:Tetrin place of prcK was integrated into the chromosome of L. paracasei HD1.7 by homologous recombination. One strain that grew only on plates with tetracycline but not on plates with ampicillin was selected. The PCR amplification and endonuclease digestion analysis indicated that plasmid pKLKRT was successfully constructed and introduced into L. paracasei HD1.7. The prcK gene deletion mutant was confirmed by PCR amplification. In conclusion, a prcK gene deletion mutant of L. paracasei HD1.7 has been successfully constructed by homologous recombination, which will lay the basis for understanding the molecular mechanism of the quorum sensing-related genes in L. paracasei HD1.7.
Key words:Lactobacillus paracasei; bacteriocin; histidine protein kinase; homologous recombination
DOI:10.7506/spkx1002-6630-201712003
中图分类号:Q935
文献标志码:A
文章编号:1002-6630(2017)12-0015-06
引文格式:岳元春, 王洋, 由田, 等. 同源重组法构建副干酪乳杆菌组氨酸蛋白激酶基因缺失突变株[J]. 食品科学, 2017, 38(12):15-20.
DOI:10.7506/spkx1002-6630-201712003. http://www.spkx.net.cn
YUE Yuanchun, WANG Yang, YOU Tian, et al. Construction of prcK gene deletion mutant of Lactobacillus paracasei by homologous recombination[J]. Food Science, 2017, 38(12): 15-20. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201712003. http://www.spkx.net.cn
收稿日期:2016-09-13
基金项目:国家自然科学基金面上项目(31470537;31570492);黑龙江省高等学校科技创新团队(农业微生物发酵技术)项目(2012td009)
作者简介:岳元春(1991—),女,硕士,研究方向为微生物资源挖掘与利用。E-mail:yueyuanchungreat@163.com
*通信作者:葛菁萍(1972—),女,教授,博士,研究方向为微生物生态学及微生物遗传育种。E-mail:gejingping@126.com