表1 PCR扩增引物
Table 1 Primers used for PCR amplification

注:加下划线表示重叠核苷酸序列;加粗并加下划线部分表示限制性内切酶位点;斜体加下划线部分表示加入6 个组氨酸。
崔 青,钱炳俊,姚晓敏,季顺利,鲁飞凤,吴 静,张建华*
(上海交通大学农业与生物学院,陆伯勋食品安全研究中心,上海 200240)
摘 要:纳豆激酶由纳豆芽孢杆菌(Bacillus subtilis natto)的aprN基因编码,在体内外具有很强的溶解纤维蛋白活性。利用聚合酶链式反应(polymerase chain reaction,PCR)技术扩增B. subtilis natto的aprN基因,并依据B. subtilis的密码子偏好性优化了起始30 个氨基酸的密码子,构建了重组表达质粒pHT01-aprN。经限制性酶酶切、PCR扩增和测序验证了其编码的正确性。通过电击法将含有强启动子的pHT01-aprN导入B. subtilis,利用氯霉素抗性筛选获得B.s 168/pHT01-aprN工程菌。经IPTG诱导表达,摇瓶发酵培养最高酶活力为(289.00±3.42) U/mL,是野生菌的3.9 倍,酶活力表达稳定性良好。
关键词:纳豆激酶;aprN基因;枯草芽孢杆菌;基因工程菌;酶活力
纳豆是利用纳豆芽孢杆菌(Bacillus subtilis natto)发酵黄豆制成的豆制品,是日本的一种传统食品,因其含有的纳豆激酶(nattokinase,NK)具有抗血栓作用深受人们喜爱,已发展为保健食品。NK是由B. subtilis natto产生的一种丝氨酸蛋白酶[1],除在体内外有强烈的纤维蛋白溶解活性外,还有促进血液流动、防血小板凝聚和降血压等功效[2-3]。1980年,须见洋行博士首次发现NK[4],其分子质量为27 kD,等电点为8.6±0.3。血栓是引起的心血管疾病的风险因子之一,严重危害人类健康[5-6]。目前,溶栓剂包括注射类降解药物(尿激酶、链激酶和组织型纤维蛋白原激活剂)和口服类的类组织纤维蛋白酶的蛋白(NK和蚓激酶[7])两大类。注射类降解药物高度依赖体内组织纤维蛋白酶原的水平,存在一定毒性,半衰期短且成本高;而NK属于天然发酵产物,安全性高,具有体内效应时间长、价格低廉、有预防作用及易于规模化生产等优点,其临床应用已得到美国食品药品监督管理局认可。
提高NK工业化生产对治疗血栓疾病有重要意义。采用优化启动子和信号肽、选用蛋白酶缺失的菌株以降低NK降解或者直接改造NK基因序列等技术手段可以达到提高NK产量的目的[8-15]。例如Wu Shuming等[8]利用枯草芽孢杆菌(Bacillus subtilis)作为宿主菌构建了NK基因工程菌,实现了其可溶性表达并通过优化启动子使其酶活力增加了136%。其他细菌,例如大肠杆菌(Escherichia coli)[9-10]、地衣芽孢杆菌[11]、乳酸链球菌[12]和酵母菌[13]等也被作为宿主菌通过基因工程发酵生产NK[14]。但这些技术也存在一些需要解决的问题,如在E. coli中表达出没有活性的包涵体;在其他工程菌中NK虽然能够可溶性表达,但酶产量低[15-17],纯化回收过程复杂[18],因此工程菌的构建策略有待进一步优化,选择合适宿主菌株和采用恰当的基因工程手段构建产NK基因工程菌株成为研究了热点。
Nishito等[19]已经证明,B. subtilis natto属于B. subtilis的一个亚种。B. subtilis无致病性、具备包含转录、翻译、蛋白质折叠和分泌等机制,常常作为工程菌株构建载体。本课题组前期实验发现B. subtilis 168含有NK基因序列,但不能表达有活性的NK。基于E. coli-B. subtilis穿梭质粒的表达载体pHT01可以在B. subtilis中高效表达重组外源蛋白。但每种生物都表现出某种程度的密码子利用的差异或偏爱,如E. coli、酵母和果蝇中编码丰度高的蛋白质的基因明显避免低利用率的密码子[20]。因此,重组蛋白的表达可能受密码子利用的影响。本研究采用分子生物学方法,依据B. subtilis的基因密码子偏好性,优化aprN基因编码前30 个氨基酸的序列,以pHT01为表达载体,成功构建B.s 168/pHT01-aprN,经摇瓶发酵培养最高酶活力可达(289.00±3.42)U/mL,酶活力表达稳定性良好。NK中加入6 个组氨酸标记,可采用镍柱亲和层析进行纯化。本实验为NK基因工程进一步研究提供参考依据。
1.1 材料与试剂
1.1.1 菌株与载体
B. subtilis 168菌株为江南大学陈卫教授惠赠;B. subtilis natto和E. coli DH5α为本实验室保藏;pMD19-T Simple载体 大连TaKaRa公司;pHT01 美国Invitrogen公司。
1.1.2 试剂
限制性内切酶BamHⅠ、EcoRⅤ、HindⅢ和SmaⅠ大连TaKaRa公司;氯霉素、氨苄卡那霉素、蛋白胨、酵母提取物、山梨醇、甘露醇、海藻糖 英国Oxoid公司;D-Val-Leu-Lys-对硝基苯胺 上海鼓臣生物技术有限公司。
1.1.3 培养基
LB液体培养基:蛋白胨10 g/L、氯化钠10 g/L、酵母提取物5 g/L。
LB固体培养基:LB液体培养基添加15 g/L琼脂糖。
发酵培养基A:葡萄糖10 g/L、酵母提取物10 g/L、K2HPO4·3H2O 1 g/L、MgSO4·7H2O 0.5 g/L、1%甘油、2,6-吡啶二羧酸1 mmol/L,pH 7.0~8.0。
发酵培养基B:葡萄糖20 g/L、酵母提取物20 g/L、K2HPO4·3H2O 2 g/L、MgSO4·7H2O 1 g/L、2%甘油、2,6-吡啶二羧酸1 mmol/L,pH 7.0~8.0。
1.1.4 引物的设计与合成
根据GenBank上收录NK基因序列(GI:608796180),依据宿主菌B. subtitle 168密码子偏好性(http://www.kazusa.or.jp/codon/cgi-bin/showcodon. cgi?species=1423),利用生物软件Oligo设计引物(表1),扩增aprN基因并优化它的前30个氨基酸密码子。引物由生工生物工程(上海)股份有限公司合成。
表1 PCR扩增引物
Table 1 Primers used for PCR amplification
注:加下划线表示重叠核苷酸序列;加粗并加下划线部分表示限制性内切酶位点;斜体加下划线部分表示加入6 个组氨酸。
1.2 仪器与设备
聚合酶链式反应(polymerase chain reaction,PCR)仪、微量台式离心机 德国Eppendrof公司;电转仪英国Biochrom公司;聚丙烯酰胺凝胶电泳仪 美国Bio-Rad公司。
1.3 方法
1.3.1 扩增与优化aprN基因
将保藏的B. subtilis 168菌株活化后,按照2%的接种量接种于LB培养基中,37 ℃、200 r/min培养12 h。目的基因的克隆采用重叠PCR的方法[21],即以菌液为第1次PCR扩增模版,将凝胶电泳验证后的胶回收产物作为第2次PCR扩增模版,再以此次胶回收产物为模版进行第3次PCR扩增。引物依次为F1-R1、F2-R1和F3-R2His,扩增并优化源于B. subtilis natto的aprN基因。
1.3.2 目的基因克隆和测序
试剂盒回收并纯化第3次PCR扩增产物,产物通过TA克隆连到质粒pMD19-T。pMD19-T载体连接体系(10 μL):DNA 4.5 μL、pMD19-T 0.5 μL、缓冲溶液Ⅰ5 μL,4 ℃过夜连接。通过热激转化方法将连接产物导入E. coli感受态中,涂布Amp抗性平板(终质量浓度100 μg/mL)。提取阳性克隆菌落质粒DNA,通过限制性内切酶酶切图谱分析和PCR产物测序验证,将携带pMD19-T∶aprN的质粒菌株置于甘油管-80 ℃保存。
1.3.3 表达载体的构建及验证
用限制性内切酶EcoRⅤ和BamHⅠ酶切pMD19-T∶aprN,切下的aprN基因片段插入pHT01的SmaⅠ和BamHⅠ酶切位点之间(多克隆位点位于启动子和终止子之间),其中EcoRⅤ和SmaⅠ均为平末端酶,经T4连接酶可以识别并连接。通过热激转化方法将连接产物导入E. coli感受态中,涂布Amp抗性平板(终质量浓度100 μg/mL)筛选转化子。提取阳性克隆菌落质粒DNA,通过限制性内切酶酶切图谱分析和PCR产物测序验证后,将携带pHT01:aprN的质粒菌株置于甘油管-80 ℃保存。1.3.4 B.s 168/pHT01-aprN的构建及鉴定
B. subtilis 168的感受态细胞制备和电击转化均参考Xue Gangping[22]的方法。随机挑取氯霉素抗性平板上长出的单菌落,提取质粒DNA,PCR产物测序验证,将携带pHT01-aprN的质粒菌株置于甘油管-80 ℃保存。
1.3.5 发酵培养
取甘油管中B.s 168/pHT01-aprN、B. subtilis natto野生菌和B. subtilis168宿主菌分别以2%接种量接种于LB培养基(含5 μg/mL氯霉素)中,200 r/min、37 ℃过夜培养。以2%接种量将B.s 168/pHT01-aprN种子液接种于25 mL发酵培养基A(含5 μg/mL氯霉素)中,37 ℃、200 r/min摇床培养,至OD600nm在0.8~1.0之间,降温至25 ℃,接入终浓度为0.5 mmol/L异丙基-β-D-硫代半乳糖苷(isopropyl β-D-1-thiogalactopyranoside,IPTG)。对照组以2%接种量分别将B. subtilis natto野生菌和B. subtilis 168宿主菌种子液接种于25 mL发酵培养基A中,37 ℃、200 r/min摇床培养。在不同发酵时间同时取3 种菌液测定OD600nm和NK酶活力。
1.3.6 NK活力测定
NK活力分析采用四肽底物法[23]。首先制作对硝基苯胺的标准曲线:配制对硝基苯胺样品溶液,浓度分别为0.02、0.03、0.04、0.06、0.08 mmol/L,在405 nm波长处测定不同浓度硝基苯胺样品溶液的OD值,以水作为空白对照。取发酵液于4 ℃、8 000 r/min离心1 min,将上清液稀释成不同浓度作为检测样品液。检测方法在文献[24]基础上加以改进:取100 µL发酵上清液稀释一定倍数与50 µL终浓度为0.5 mmol/L的四肽底物(D-Val-Leu-Lys-对硝基苯胺)混合并在37 ℃水浴下反应1 min,加入75 µL 50%冰醋酸溶液终止反应,在405 nm波长处测定反应液OD值。NK活力单位被定义为每分钟释放1 nmol对硝基苯胺的NK含量。实验重复3 次,每次3 个平行。
1.3.7 酶活力的影响因素分析
探究2,6-吡啶二羧酸、IPTG、诱导温度、接种量对酶活力的影响,以培养基A为发酵培养基,2,6-吡啶二羧酸浓度设置为0.5、1.0、2.0 mmol/L;IPTG浓度设置为:0.2、0.5、1.0 mmol/L;诱导温度设置为25、30、37 ℃;接种量设置为1%、2.5%、5%和7.5%。为提高NK酶活力,确定培养基成分后再将碳源、氮源和无机盐浓度提高1 倍。以B. subtilis natto野生菌和宿主菌B. subtilis168作对照。
1.4 数据统计分析
数据处理采用SAS 8.2统计软件进行方差分析,采用邓肯氏多重(Duncan’s multiple range test)比较分析差异显著性。
2.1 aprN基因的克隆和表达载体构建
2.1.1 aprN基因的克隆
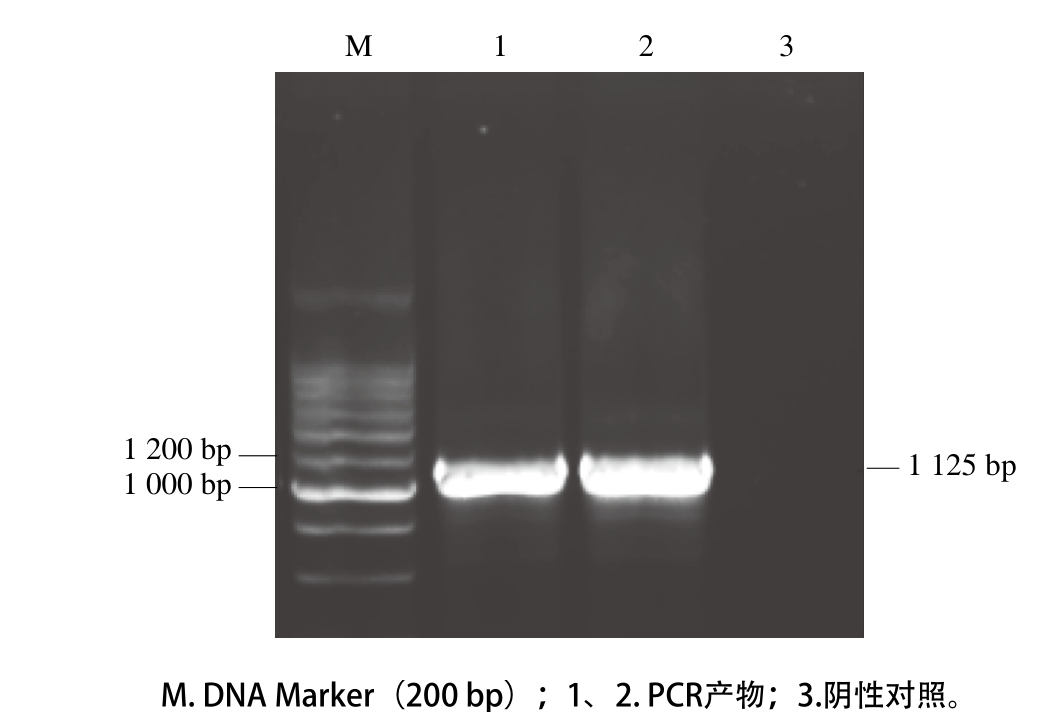
图1 琼脂糖凝胶电泳鉴定PCR扩增产物
Fig. 1 Identification of PCR products by agarose gel electrophoresis
利用设计的引物依次进行PCR扩增,琼脂糖凝胶电泳鉴定第3次PCR产物,在1 100 bp附近有十分明显的DNA扩增带,与预期aprN基因长度1 125 bp吻合(图1)。PCR产物序列结果与NCBI上序列对比,同源性达99%。

图 2aprN基因前30 个氨基酸密码子优化前后的序列比对
Fig. 2 Sequence comparison of the codons encoding the first 30 amino acids of aprN gene before and after optimization
重组蛋白的表达可能受密码子利用的影响,为提高B. subtilis 168中NK的翻译速率,依据B. subtilis的密码子偏好性,优化了NK前30 个氨基酸密码子,密码子优化前后的序列如图2所示。经限制性内切酶EcoRⅤ和BamHⅠ双酶切,鉴定插入片段与预期一致,如图3所示,1 100 bp左右的条带是目的基因aprN,克隆载体构建成功。

图3 pMD19-T:aprN经EcoR Ⅴ/BamHⅠ酶切电泳图
Fig. 3 Electrophoretogram of plasmid pMD19-T:aprN digested by EcoR Ⅴ and BamH Ⅰ
2.1.2 pHT01-aprN重组质粒构建
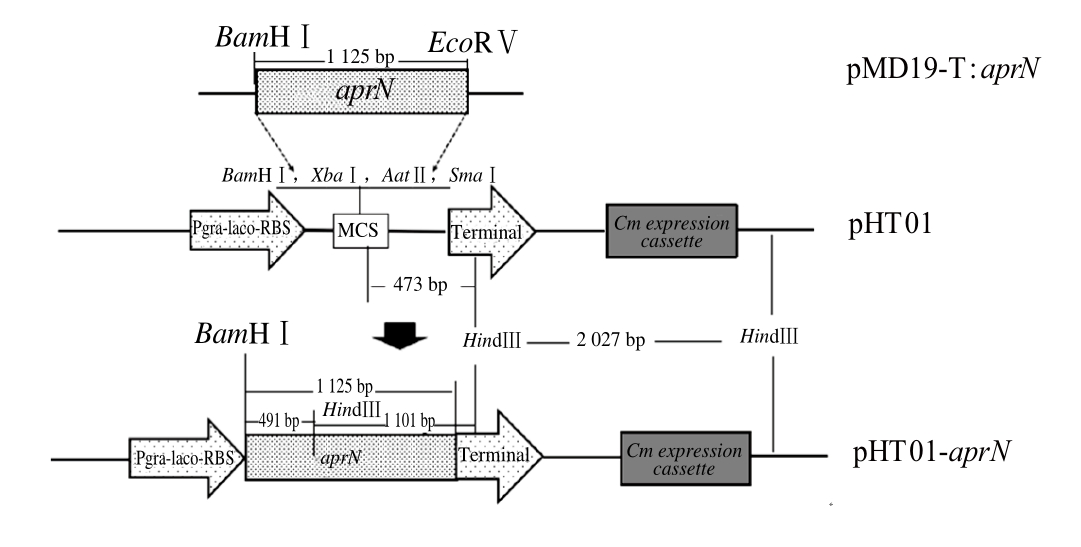
图4 pHT01-aprN重组质粒图
Fig. 4 Schematic diagram of pHT01-aprN
pHT01可在细胞质中高水平表达重组蛋白,其携带的启动子是基于强σA-依赖性启动子的B. subtilis groE操纵子,通过添加乳糖操纵子改造而成的一种高效可控的(IPTG诱导的)启动子。该载体还插入了一个高效SD序列以及一个多克隆位点(BamHⅠ、XbaⅠ、AatⅡ、SmaⅠ)。aprN基因信号肽编码区域与SD序列融合以此获得分泌的重组蛋白。本实验尝试采用pHT01载体构建表达质粒,有望提高NK在B. subtilis 168中翻译速率并实现胞外表达。
经限制性内切酶EcoRⅤ/BamHⅠ从pMD19-T:aprN切下的aprN基因片段插入pHT01的多克隆酶切位点BamHⅠ和SmaⅠ之间,构建重组质粒pHT01-aprN,如图4所示。
由图4可知,pHT01-aprN重组质粒的启动区域包含groE启动子(gra)、lacO操纵子和RBS序列,阻遏蛋白LacI与LacO结合阻遏转录起始,乳糖的类似物IPTG可以和LacI产物结合,使其构象改变离开LacO,从而激活转录。插aprN基因长1 125 bp,该质粒含有氯霉素表达区域,可以氯霉素作为重组质粒筛选标记。BamHⅠ和HindⅢ酶切位点用于后续鉴定。
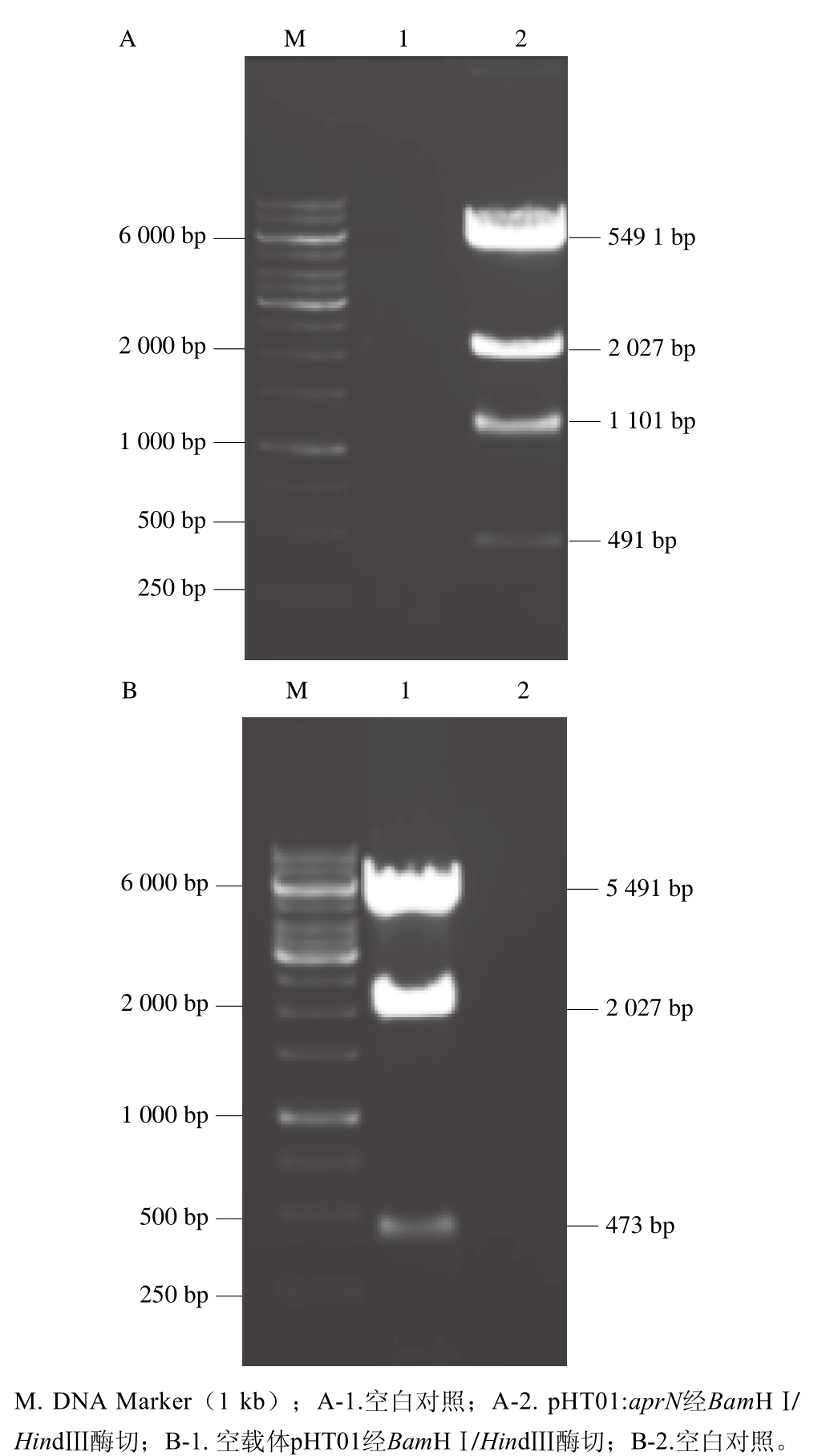
图5 pHT01-aprN经BamHⅠ/HindⅢ酶切电泳图
Fig. 5 Electrophoretogram of plasmid pHT01-aprN digested by BamHⅠand HindⅢ
pHT01-aprN经限制性内切酶BamHⅠ和HindⅢ酶切后,切得条带分别为491、1 101、2 027 bp(图5A);图5B显示pHT01质粒经相同酶切后,切得条带分别为473 bp和2 027 bp。切得条带大小与预期结果一致(图4),表明图5A-2泳道的转化子应为阳性克隆,该菌株中的质粒pHT01上已成功插入aprN基因片段,携带pHT01-aprN重组质粒的基因工程菌B.s 168/pHT01-aprN构建成功。
2.1.3 B.s 168/pHT01-aprN工程菌鉴定
在所有革兰氏阳性菌中,B. subtilis载体因下列原因尤为引人瞩目[24]:1)一般认为是安全的有机体;2)可胞外分泌蛋白;3)具备包含转录、翻译、蛋白质折叠和分泌机制,可对其进行遗传操作和大规模发酵;4)细胞壁的组成简单,只含有肽聚糖和磷壁酸,因此在分泌的蛋白质产品中不会混杂有胞被内毒素。
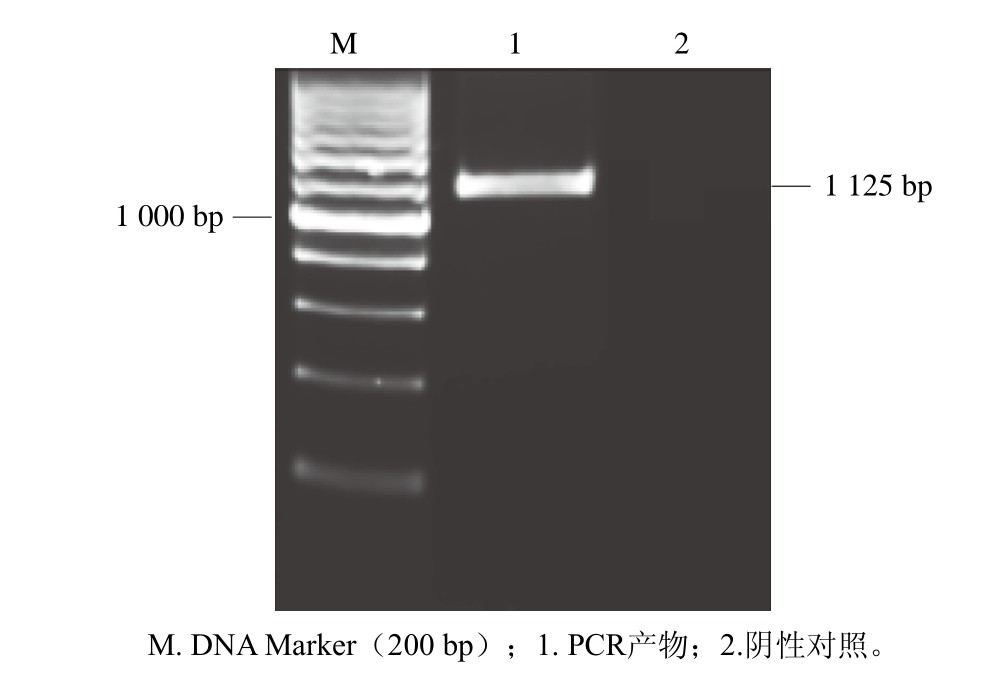
图6 琼脂糖凝胶电泳鉴定阳性菌落PCR扩增产物
Fig. 6 Identification of PCR products by agarose gel electrophoresis
pHT01-aprN重组质粒电转入B. subtilis 168,阳性菌落PCR扩增图谱如图6所示,在1 100 bp附近有明显的特异性扩增条带,与目的基因相符,经测序验证正确,证明携带pHT01-aprN重组质粒的工程菌B.s 168/pHT01-aprN构建成功。
2.2 NK酶活力影响因素分析
2.2.1 酶活力测定标准曲线
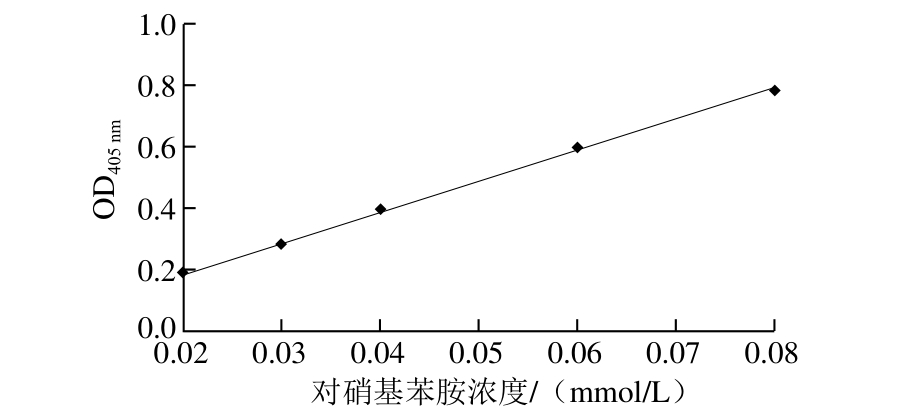
图7 NK活力检测标准曲线
Fig. 7 Standard curve for NK activity measurement
四肽底物法是测定NK酶活力的常用方法之一,主要是考察NK水解酰胺键的能力检测酶活力,具有检测快速、方便、灵敏度高的特点。NK活力检测标准曲线如图7所示,其回归方程为y=10.002 0x-0.008 9,R2为0.999 1,有良好的线性关系。
2.2.2 IPTG对NK酶活力的影响

图8 IPTG浓度对细胞生长的影响
Fig. 8 Effects of IPTG concentration on cell growth
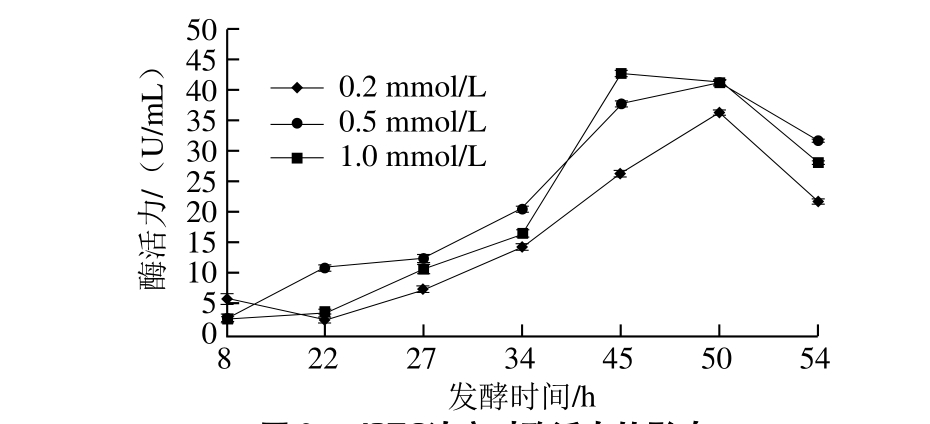
图9 IPTG浓度对酶活力的影响
Fig. 9 Effects of IPTG concentration on enzyme activity
Wang等[25]优化发酵培养基的实验证明:葡萄糖、K2HPO4·3H2O和MgSO4·7H2O对B. subtilis natto产NK起关键作用;Berenjian等[23]证明甘油促进NK表达。本实验在文献基础上[22,24-28],以酵母提取物为氮源进行发酵。
由图8、9可知,IPTG浓度对工程菌细胞生长影响不显著(P>0.05)。发酵45、50 h,在0.5 mmol/L和 1 mmol/L浓度下酶活力差异不显著(P>0.05),但两者与0.2 mmol/L相比差异显著;发酵54 h,3 个浓度差异不显著(P>0.05),IPTG浓度为0.5 mmol/L时酶活力相对最高。乳糖的类似物IPTG起诱导表达作用的同时还存在一定毒性,需控制在最适范围内既提高表达量又不影响菌体生长,实验选用IPTG浓度为0.5 mmol/L。
2.2.3 2,6-吡啶二羧酸对NK酶活力的影响
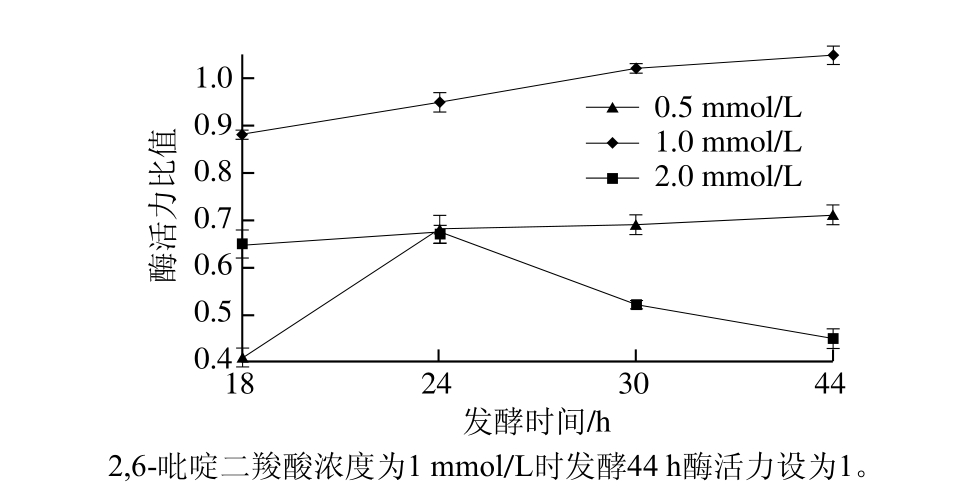
图10 吡啶二羧酸浓度对酶活力的影响
Fig. 10 Effects of pyridine dicarboxylic acid concentration on enzyme activity
2,6-吡啶二羧酸是存在于芽孢杆菌中的抗菌剂,它可提高渗透压,有利于维持酶的构象,浓度适当可提高NK活力[26],本实验探究2,6-吡啶二羧酸浓度对酶活力的影响。由图10可知,不同2,6-吡啶二羧酸浓度在各时间段对酶活力影响差异显著(P<0.05)。浓度从0.5 mmol/L增大到1.0 mmol/L时,主要起促进酶活力表达的作用,酶活力升高。浓度再增加到2.0 mmol/L后起抑制作用,酶活力降低。最终确定2,6-吡啶二羧酸最佳浓度为1 mmol/L。
2.2.4 发酵液浓度对NK酶活力的影响

图11 诱导温度和培养基浓度对细胞生长的影响
Fig. 11 Effects of medium concentration and temperature on cell growth
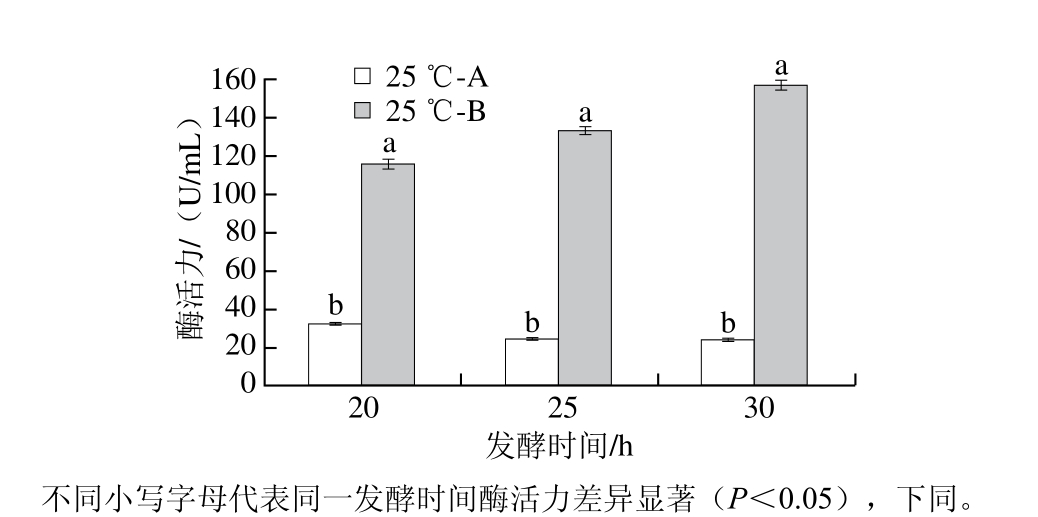
图12 25 ℃诱导温度下培养基浓度对酶活力的影响
Fig. 12 Effects of medium concentration on enzyme activity when the induction temperature was 25 ℃
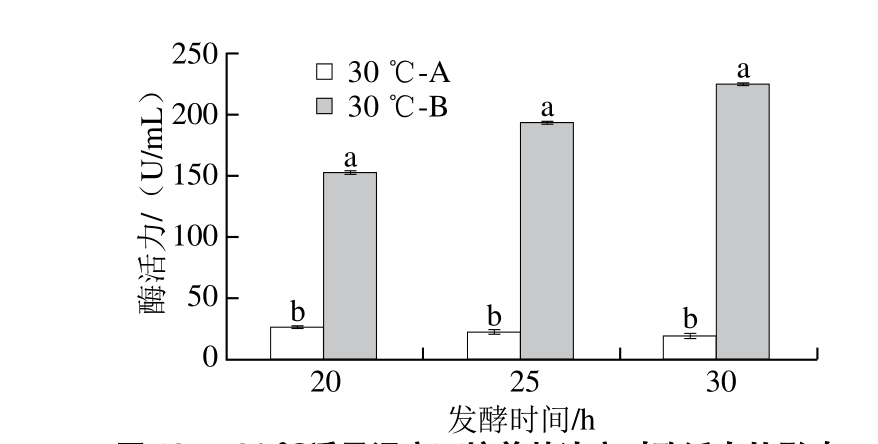
图13 30 ℃诱导温度下培养基浓度对酶活力的影响
Fig. 13 Effects of medium concentration on enzyme activity when the induction temperature was 30 ℃
确定IPTG和2,6-吡啶二羧酸添加量后,为提高NK表达量和提高菌体浓度,将发酵培养基浓度提高1 倍,结果如下:由图11、12所示,在25 ℃条件下,培养基浓度对菌体量和酶活力的影响极为显著(P<0.01),培养基浓度增加可促进细菌生长和酶活力提高。其中发酵20 h时,B的菌体浓度和NK酶活力分别是A的3.3 倍和3.6 倍;25 h时,B的菌体浓度和NK酶活力分别是A的4.8 倍和5.4 倍;而在30 h时,比值分别为4.4 倍和6.6 倍。30 h时,酶活力最高为(156.00±2.04)U/mL。
由图11和13所示,在30 ℃诱导温度下,不同培养基培养的工程菌OD600nm和酶活力存在显著差异(P<0.05),其中发酵20 h时,以B为发酵培养基的菌体浓度和NK酶活力分别是A的3.3倍和5.9倍;25 h时,比值分别为的2.8、8.5 倍;而在30 h时,比值分别为1.4 、11.5 倍。30 h时,酶活力最高为(224.00±3.08)U/mL。
在相同发酵培养基、两个不同温度下,菌体量和酶活力差异不显著,但30 ℃菌体量和酶活力表达均较高,培养基浓度增加(发酵培养基B)提高菌体浓度,是酶活力增加的重要原因。同时,在相同菌体浓度下,酶的表达量有所提高。在发酵培养基B中培养时,菌体浓度的增加主要发生在25 h以前,而酶活力在此后还会增加,说明生长和产酶并不同步。
2.2.5 接种量对NK酶活力的影响
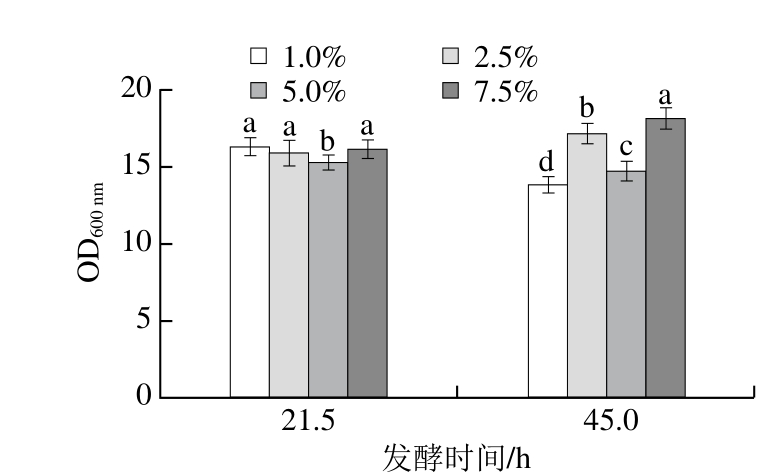
图14 接种量对细胞生长的影响
Fig. 14 Effects of inoculum concentration on cell growth
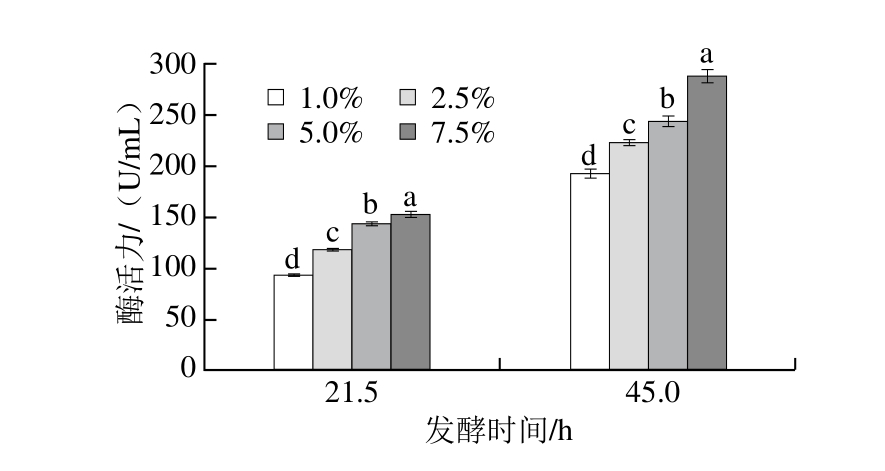
图15 接种量对酶活力的影响
Fig. 15 Effects of inoculum concentration on enzyme activity
由图14可知,发酵21.5和45 h,不同接种量引起的菌体量差异不显著,其中在20 h左右接种量为5% 时菌体量较低,接种量为2.5%和7.5%菌体量较大。由图15可知,发酵21.5 h,不同接种量引起的酶活力差异显著(P<0.05),随着发酵45 h,酶活力差异不显著,但接种量越大,酶活力越高。其中当接种量为7.5%时酶活力平均最高值为(289.26±3.42)U/mL。
采用相同的培养基摇瓶发酵,未检测出宿主菌B. subtilis 168中的酶活力,B. subtilis natto酶活力为(74.32±0.69)U/mL,工程菌B.s 168/pHT01-aprN酶活力是野生菌B. subtilis natto的3.9 倍,可见B.s 168/pHT01-aprN在很大程度上提高了酶活力。Wei Xuetuan等[27]构建的工程菌NK酶活力(工业法33.86 FU/mL)是原宿主菌WX-02的2.3 倍,在相同的酶活力测定方法(四肽底物法)下,B.s 168/pHT01-aprN摇瓶培养酶活力是Berenjian等[23]报道的野生菌NK酶活力(23.48 U/mL)的12.2 倍,是Kim等[28]报道的(13.5 U/mL)21.5 倍,大大提高NK胞外表达水平。Berenjian等[23]报道优化培养基进行高密度发酵酶活力为587 U/mL,比摇瓶培养的野生菌酶活力(23.48 U/mL)提高25 倍;Cho等[29]高密度发酵的NK酶活力是摇瓶培养的5.4 倍,补料发酵再提高2.1 倍;Kwon等[30]高密度发酵的NK酶活力是摇瓶培养的5.0 倍,补料发酵再提高4.3 倍。以上文献报道的结果对下一步进行高密度发酵提供有力支持。
本实验采用pHT01载体,依据B. subtilis 密码子的偏爱性,优化编码NK前30 个氨基酸的密码子,实现优化的aprN基因在B. subtilis 168中表达。通过酶活力比较,最终确定发酵培养基条件为:培养基成分(葡萄糖20 g/L、酵母提取物20 g/L、K2HPO4·3H2O 2 g/L、MgSO4·7H2O 1 g/L、甘油2%,pH 8.0)、2,6-吡啶二羧酸1 mmol/L、IPTG 终浓度0.5 mmol/L、接种量7.5%、发酵诱导温度30 ℃,该条件下NK最高酶活力为(289.26±3.42) U/mL,胞外NK酶活力为相同培养基培养下的野生菌的3.9 倍,酶活力表达稳定性良好。NK中加入了6 个组氨酸片段,有助于后续利用镍柱亲和层析进行纯化。后续研究高密度发酵后对酶活力表达的影响。
参考文献:
[1] SUMI H, HAMADA H, TSUSHIMA H, et al. A novel fibrinolytic enzyme (nattokinase) in the vegetable cheese Natto: a typical and popular soybean food in the Japanese diet[J]. Experientia, 1987, 43(10): 1110-1111. DOI:10.1016/0268-9499(88)90502-4.
[2] FUJITA M, OHNISHI K, TAKAOKA S, et al. Antihypertensive effects of continuous oral administration of nattokinase and its fragments in spontaneously hypertensive rats[J]. Biological & Pharmaceutical Bulletin, 2011, 34(11): 1696-1701. DOI:10.1248/bpb.34.1696.
[3] JANG J y, KIM T S, CAI J, et al. Nattokinase improves blood flow by inhibiting platelet aggregation and thrombus formation[J]. Laboratory Animal Research, 2013, 29(4): 221-225. DOI:10.5625/ lar.2013.29.4.221.
[4] BEL L. Purification and serology of a Bacilliform virus associated with banana streak disease[J]. Hytopathology, 1986, 76(10): 995-999. DOI:10.1094/Phyto-76-995.
[5] 林超, 刘兆国, 钱星, 等. 丹酚酸B在心血管疾病中药理作用研究进展[J]. 中国药理学通报, 2015, 31(4): 449-452. DOI:10.3969/ j.issn.1001-1978.2015.04.002.
[6] WEI X, LUO M, XIE Y, et al. Strain screeung, fermentation, separation and encapsulation for production of nattokinase functional food[J]. Applied Biochemistry and Biotechnology, 2012, 168(7): 1753-1764. DOI:10.1007/s12010-012-9894-2.
[7] PENG Y, HUANG Q, ZHANG R H, et al. Purification and characterization of a fibrinolytic enzyme produced by Bacillus amyloliquefaciens DC-4 screened from douchi, a traditional Chinese soybean food[J]. Comparative Biochemistry & Physiology Part B Biochemistry & Molecular Biology, 2003, 134(1): 45-52. DOI:10.1016/S1096-4959(02)00183-5.
[8] WU S M, FENG C, ZHONG J, et al. Enhanced production of recombinant nattokinase in Bacillus subtilis by promoter optimization[J]. World Journal of Microbiology and Biotechnology, 2011, 27(1): 99-106. DOI:10.1007/s11274-010-0432-5.
[9] CHIANG C J, CHEN H C, CHAD Y P, et al. Efficient system of artificial oil bodies for functional expression and purification of recombinant nattokinase in Escherichia coli[J]. Journal of Agricultural & Food Chemistry, 2005, 53(12): 4799-4804. DOI:10.1021/jf050264a.
[10] 黄磊, 谢玉娟, 李申, 等. 纳豆激酶基因的克隆及其在大肠杆菌和枯草芽孢杆菌中的表达[J]. 食品科学, 2007, 28(5): 199-202. DOI:10.3321/j.issn:1002-6630.2007.05.046.
[11] WEI X, ZHOU y, CHEN J, et al. Efficient expression of nattokinase in Bacillus licheniformis: host strain construction and signal peptide optimization[J]. Industrial Microbiology & Biotechnology, 2016, 42(2): 287-295. DOI:10.1007/s10295-014-1559-4.
[12] LIANG X B, ZHANG L X, ZHONG J, et al. Secretory expression of a heterologous nattokinase in Lactococcus lactis[J]. Applied Microbiology and Biotechnology, 2007, 75(1): 95-101. DOI:10.1007/ s00253-006-0809-4.
[13] 敬俊锋, 陈斌, 李莹, 等. 纳豆激酶基因的克隆及其在毕赤酵母中的表达[J]. 生物学杂志, 2011, 28(5): 55-60. DOI:10.3969/ j.issn.2095-1736.2011.05.055.
[14] 季顺利, 蔡俊秀, 崔青, 等. 纳豆激酶的制备及其改性研究进展[J]. 中国酿造, 2016, 35(6): 6-10. DOI:10.3969/j.issn.1004-5503.2008.12.030.
[15] PENG y, yANG X, ZHANG y, et al. Microbial fibrinolytic enzymes: an overview of source, production, properties, and thrombolytic activity in vivo[J]. Applied Microbiology and Biotechnology, 2005, 69(2): 126-132. DOI:10.1007/s00253-005-0159-7.
[16] CEREGHINO J L, CREGG J M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris[J]. FEMS Microbiology Reviews, 2000, 24(1): 45-66. DOI:10.1111/j.1574-6976.2000.tb00532.x.
[17] LIANG X, JIA S, SUN Y, et al. Secretory expression of nattokinase from Bacillus subtilis YF38 in Escherichia coli[J]. Molecular Biotechnology, 2007, 37(3): 187-194. DOI:10.1007/s12033-007-0060-y.
[18] BORAH D, SHAHIN L, SANGRA A, et al. Production, purification and characterization of nattokinase from Bacillus subtilis, isolated from tea garden soil samples of Dibrugarh, Assam[J]. Asian Journal of Pharmaceutical & Clinical Research, 2012, 5(3): 124-125. DOI:10.1023/A:1015252607118.
[19] NISHITO Y, OSANA Y, HACHIYA T, et al. Whole genome assembly of a natto production strain Bacillus subtilis natto from very short read data[J]. BMC Genomic, 2010, 11: 243. DOI:10.1186/1471-2164-11-243.
[20] 唐晓芬, 陈莉, 马玉韬. 密码子使用偏性量化方法研究综述[J].基因组学与应用生物学, 2013, 32(5): 660-666. DOI:10.3969/ gab.032.000660.
[21] BING Junqian, SHEN Huifeng, XIONG Jingjing, et al. Expression and purification of the synthetic preS1 gene of Hepatitis B Virus with preferred Escherichia coli codon preference[J]. Protein Expression and Purification, 2006, 48(1): 74-80. DOI:10.1016/j.pep.2005.11.024.
[22] XUE G P, JOHNON J S, DALRYMPLE B P. High osmolarity improves the electro-transformation efficiency of the gram-positive bacteria Bacillus subtilis and Bacillus licheniformis[J]. Journal of Microbiological Methods, 1999, 34(3): 183-191. DOI:10.1016/S0167-7012(98)00087-6.
[23] BERENJIAN A, MAHANAMA R, KAVANAGH J, et al. Nattokinase production: medium components and feeding strategy studies[J]. Chemical Industry & Chemical Engineering Quarterly, 2014, 20(41): 541-547. DOI:10.2298/CICEQ130928037B.
[24] 余晓霞, 田健, 刘晓青, 等. 枯草芽孢杆菌表达系统及其启动子研究进展[J]. 生物技术通报, 2015, 31(2): 35-44. DOI:10.13560/j.cnki. biotech.bull.
[25] WANG J K, CHIU H H, HSIEH C S. Optimization of the medium components by statistical experimental methods to enhance nattokinase activity[J]. Fooyin Journal of Health Sciences, 2009, 1(1): 21-27. DOI:10.1016/S1877-8607(09)60004-7.
[26] SHIORI I, TADANORI O, HIROyUKI S. Activation of fibrinolysis (nattokinase) induced by dipicolinic acid and related compounds[J]. Food Science & Technology Research, 2006, 12(2): 152-155. DOI:10.3136/fstr.12.152.
[27] WEI X T, ZHOU Y H, CHEN J B, et al. Efficient expression of nattokinase in Bacillus licheniformis: host strain construction and signal peptide optimization[J]. Journal of Industrial Microbiology & Biotechnology, 2015, 42(2): 287-295. DOI:10.1007/s10295-014-1559-4.
[28] KIM W, CHOI K, KIM Y. Purification and characterization of a fibrinolytic enzyme produced from Bacillus sp. strain CK 11-4 screened from Chungkook-Jang[J]. Applied and Environmental Microbiology, 1996, 67(2): 2482-2488. DOI:0099-2240/96/$04.0010.
[29] CHO Y H, SONG J Y, KIM K M, et al. Production of nattokinase by batch and fed-batch culture of Bacillus subtilis[J]. New Biotechnology, 2010, 27(4): 341-346. DOI:10.1016/j.nbt.2010.06.003.
[30] KWON E Y, KIM K M, KIM M K, et al. Production of nattokinase by high cell density fed-batch culture of Bacillus subtilis[J]. Bioprocess and Biosystems Engineering, 2011, 34(7): 789-793. DOI:10.1007/ s00449-011-0527-x.
Construction of Genetically Engineered Strain for Nattokinase Production and Enzyme Activity Analysis
CUI Qing, QIAN Bingjun, YAO Xiaomin, JI Shunli, LU Feifeng, WU Jing, ZHANG Jianhua*
(Bor S. Luh Food Safety Research Center, School of Agriculture and Biology, Shanghai Jiao Tong University, Shanghai 200240, China)
Abstract:Nattokinase (NK), encoded by the aprN gene of Bacillus subtilis natto, has strong fibrinolytic activity both in vitro and in vivo. In this research, the aprN gene from B. subtilis was cloned and the codons which encode the first 30 amino acids were optimized on the basis of the codon preference of B. subtilis. Recombinant vector pHT01-aprN was constructed. Through restriction enzyme digestion analysis, PCR amplification and sequencing, the subcloned gene was confirmed to be aprN. The pHT01-aprN was transformed into B. subtilis 168 by electroporation, and the engineered bacterium (B.s 168/pHT01-aprN) was isolated on LB plates containing chloramphenicol. NK expression was induced by IPTG, and the highest enzyme activity in shaking flask culture was up to (289.00 ± 3.42) U/mL with good stability, which was 3.9 times as high as that of wild-type B. subtilis natto.
Key words:nattokinase; aprN gene; Bacillus subtilis; enzyme activity; engineered bacteria
DOI:10.7506/spkx1002-6630-201714001
中图分类号:Q786
文献标志码:A
文章编号:1002-6630(2017)14-0001-08
引文格式:
崔青, 钱炳俊, 姚晓敏, 等. 纳豆激酶基因工程菌的构建及酶活力分析[J]. 食品科学, 2017, 38(14): 1-8. DOI:10.7506/ spkx1002-6630-201714001. http://www.spkx.net.cn
CUI Qing, QIAN Bingjun, YAO Xiaomin, et al. Construction of genetically engineered strain for nattokinase production and enzyme activity analysis[J]. Food Science, 2017, 38(14): 1-8. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201714001. http://www.spkx.net.cn
收稿日期:2016-10-08
基金项目:国家自然科学基金面上项目(31171737)
作者简介:崔青(1992—),女,硕士研究生,研究方向为食品微生物。E-mail:cuiqing1992@sina.com
*通信作者:张建华(1968—),男,副研究员,博士,研究方向为食品微生物。E-mail:zhangjh@sjtu.edu.cn