
图1 豆酱样品细菌总DNA的PCR-DGGE图谱
Fig. 1 DGGE atlas of bacterial community in each soybean paste sample
张 颖1,乌日娜1,2,*,孙慧君1,3,武俊瑞1,陶冬冰1,王洪玉1
(1.沈阳农业大学食品学院,辽宁 沈阳 110866;2.食品科学与技术国家重点实验室,江南大学食品学院,江苏 无锡 214122;3.辽宁省农业经济学校现代农业技术系,辽宁 锦州 121001)
摘 要:以东北传统自然发酵豆酱为研究对象,采用聚合酶链式反应-变性梯度凝胶电泳(polymerase chain reactiondenaturing gradient gel electrophoresis,PCR-DGGE)技术结合高通量测序方法分析豆酱发酵过程中细菌的多样性及动态变化。结果显示,细菌的可操作分类单元数值在发酵过程中上下波动,并在后发酵第21天时达到最大值,此时细菌数目最多;整理分析PCR-DGGE图谱鉴定结果以及细菌在属水平上的分布得出,明串珠菌属、肠球菌属、四联球菌属和乳杆菌属为豆酱样品不同发酵阶段的优势细菌菌属,其中四联球菌属波动性较大,易受内外环境的影响;屎肠球菌、明串珠菌属、绿色魏斯菌随着发酵时间的延长在减少,说明这些细菌主要参与豆酱发酵初期产酶分解物质过程,并在整个发酵过程中起到调整发酵内环境等作用。
关键词:东北传统发酵豆酱;PCR-DGGE;微生物多样性测序;优势细菌菌属;可操作分类单元
引文格式:张颖, 乌日娜, 孙慧君, 等. 豆酱不同发酵阶段细菌群落多样性及动态变化分析[J]. 食品科学, 2017, 38(14): 30-35.
DOI:10.7506/spkx1002-6630-201714005. http://www.spkx.net.cn
ZHANG Ying, WU Rina, SUN Huijun, et al. Diversity and dynamic changes of the bacterial community during fermentation of soybean paste[J]. Food Science, 2017, 38(14): 30-35. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201714005. http://www.spkx.net.cn
传统发酵豆酱是以大豆为原料,经过煮制、挤压成块、制曲、发酵而成[1],其色泽为红褐色或棕褐色,有光泽,有明显的酱香和酯香,咸淡适口,呈半流动状态[2]。东北传统豆酱的做法:将挑好洗净的东北大豆蒸煮6 h,绞碎豆粒制成6 cm×5 cm×5 cm的长方体,置于阳光下发酵2 个月待干燥后用报纸或纱布包好继续发酵1 个月成曲;洗掉成曲上的菌丝体后将其切成小块与一定比例的盐和水一并放入缸中进行后期发酵,早晚各搅拌一次直至发酵成熟[3]。豆酱作为主要发酵豆制品之一,可以助消化,促进人体造血、提高肝脏的解毒功能、降低血脂、预防癌症发生[4]。豆酱的品质与营养是通过原料和环境中的多种微生物的协同作用[8]以及利用碳源、氮源等经过一系列生化反应所产生的,但目前对传统发酵豆酱的研究中,主要集中在发酵过程中理化指标、流变学特性及质地评价、风味物质萃取的条件等[5-7]。因此深入研究传统豆酱发酵不同阶段微生物的多样性和菌群动态,掌握微生物的生长规律,为改善工业豆酱品质,发酵菌株进行系统选育,优化改良和实现产业化发展提供了理论依据。
Muzyer等[9]证明变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)技术在研究自然界微生物群落多样性及菌相差异方面具有明显的优越性,在食品领域已得到了成熟的应用,如利用聚合酶链式反应(polymerase chain reaction,PCR)-DGGE技术分析国内外自然发酵泡菜中微生物的多样性[10-12]、酒厂中成熟与退化窖泥中微生物的结构组成[13]、玉米青贮及苜蓿青贮中细菌动态发酵多样性[14]、周剑忠等[15]通过指纹图谱技术与序列分析研究发现藏灵菇中除了有大量无法培养的微生物外,还有多种酵母菌和乳酸菌的存在。PCRDGGE技术现如今已得到广泛的应用,值得注意的是当双链DNA分子在变性梯度凝胶中电泳时,为了避免DNA分子全部解链而影响结果的真实性,一般在设计上游引物时往往会加入一段30~40 bp的GC夹子[16]。它虽成功克服了基于形态学及生理生化研究技术的局限,获取了更加丰富的微生物多样性信息,但相比于当今高通量分析技术的优势,对环境中绝大部分微生物的发掘还不够全面[17]。高通量测序是指对微生物群体进行高通量测序,通过测序序列的构成分析特定环境中微生物群体的构成情况或基因的组成以及功能性,不仅为遗传信息的揭示和基因表达调控等基础生物学研究提供重要数据,而且在基因诊断和基因治疗等应用研究中也发挥着重要的作用。豆酱整个发酵过程是由多种微生物共同参与的,目前研究发现其中的主要微生物类群有霉菌[18]、酵母菌和乳酸菌[19-21],从整体上利用DGGE法结合高通量测序全面探究东北传统豆酱中微生物多样性的研究目前还鲜有报道。
本实验通过PCR-DGGE分子生物学技术结合高通量测序方法,对传统自然发酵豆酱中细菌群落进行全面系统研究,揭示不同时期的细菌菌落结构的真实状态,挖掘丰富的微生物资源并确定不同发酵阶段的优势菌群,为工业化人工接种法生产的酱类打下了良好的基础,实现传统酱类的现代化、规模化生产,提供质量安全、风味优良的豆酱以满足市场需求,为进一步深入研究和开发自然发酵豆酱中的有益菌株提供依据。
1.1 材料与试剂
实验样品:采集自两家东北地区农家(T家、Z家)利用东北传统方法制作的不同发酵阶段的豆酱样品各6 份,分别在发酵0、7、14、21、28、35 d(成品酱)进行取样(样品分别记为T1~T6、Z1~Z6),样品采集方式为随机采样。样品采集后,立即送往实验室保存于-20 ℃。
TE缓冲液、十二烷基硫酸钠(sodium dodecyl sulfate,SDS)、蛋白酶K、十六烷基三甲基溴化铵(cetyltrimethyl ammonium bromide,CTAB)、Tris饱和酚-氯仿-异戊醇(25∶24∶1,V/V)、氯仿-异戊醇(24∶1,V/V)、冰异丙醇、70%无水乙醇、10×Easy Taq Buffer、High Pure dNTPs、Easy Taq聚合酶、6×Loading buffer、DL2000 DNA Marker 北京艾比根生物有限公司。
1.2 仪器与设备
5417高速冷冻离心机 德国Eppendorf公司;Veriti96 PCR扩增仪 美国ABI公司;水浴锅、DYCP-31BN型琼脂糖凝胶电泳仪 中国深华生物技术有限公司;紫外凝胶成像及分析系统、DGGE仪 美国Bio-Rad公司;FastPrep匀浆仪 美国MP Biomedicals公司。
1.3 方法
1.3.1 样品总DNA提取
样品处理:pH 7.0、0.1 mol/L磷酸盐缓冲液悬浮8 g豆酱,用振荡器振荡均匀,样品用2 层灭菌纱布过滤,滤液经4 ℃、3 000 r/min离心10 min,收集上清液,将沉淀用pH 7.0、浓度为0.1 mol/L的磷酸盐缓冲液悬浮后离心,收集上清液,重复2 次,离心收集上清液;将所收集上清液在12 000 r/min、4 ℃条件下离心10 min,收集沉淀,将沉淀用5 mL灭菌水混匀,4 ℃、12 000 r/min离心10 min,重复2 次,收集沉淀,得到菌体沉淀。采用CTAB法进行总DNA的快速提取[22]。
1.3.2 细菌的PCR扩增
以提取的总DNA为模板,采用16S rDNA基因V3区通用引物,扩增产物片段长约250 bp。PCR体系(50 ☒L):36 ☒L ddH2O、5 ☒L 10×Easy Taq Buffer、 4 ☒L dNTPs,1.5 ☒L F338-GC,1.5 ☒L R518,0.5 ☒L的Easy Taq聚合酶及适量的ddH2O补足50 ☒L。反应参数:95 ℃预变性4 min;95 ℃ 1 min、55 ℃ 45 s、72 ℃ 1 min,30 个循环;72 ℃延伸7 min,4 ℃条件下保存。PCR产物用质量分数1%的琼脂糖凝胶电泳(100 V恒压电泳30 min)检测后-20 ℃保存备用。
1.3.3 DGGE与条带测序
采用DGGE对PCR产物进行分离。使用梯度胶制备装置,采用8%聚丙烯酰胺凝胶、35%~50%变性剂梯度。待胶完全凝固后,将胶板放入装有1×TAE电泳缓冲液的装置中,在每个加样孔中加入已和上样缓冲液混合好的PCR样品。电泳完毕后,利用GelRed染色法将凝胶染色30 min,将染好的凝胶用扫描仪成像,得到PCR-DGGE图谱,对上述扫描后的图片进行分析,标记特异性条带,并回收条带,由上海桑尼生物技术有限公司测序,测序结果进行BLAST比对鉴定。
1.3.4 高通量测序分析
1.3.4.1 可操作分类单元(operational taxonomic unit,OTU)鉴定结果统计
使用QIIME软件,调用UCLUST这一序列比对工具[23],对前述获得的高质量序列按97%的序列相似度进行归并和OTU划分,选取每个OTU中丰度最高的序列作为该OTU的代表序列。在QIIME软件中使用默认参数,通过将OTU代表序列与对应数据库的模板序列相比对,获取每个OTU所对应的分类学信息。
1.3.4.2 基于UniFrac距离的样本聚类分析
UniFrac距离就是通过比较两个群落各自独有OTU之间系统发育关系的远近,从而更全面地反映群落样本之间的相似程度[24]。使用QIIME软件对UniFrac距离矩阵分别进行UPGMA聚类分析,并使用R软件进行可视化。
1.3.4.3 属水平的分类学组成分析
使用QIIME软件,获取各样本属分类水平上的组成和丰度分布表,并通过柱状图呈现分析结果。
2.1 PCR扩增产物DGGE图谱分析
采用CTAB法提取豆酱样品总DNA,并特异扩增细菌16S rDNA V3区,其PCR-DGGE图谱如图1所示。对各条带的鉴定结果见表1。
图1 豆酱样品细菌总DNA的PCR-DGGE图谱
Fig. 1 DGGE atlas of bacterial community in each soybean paste sample
表1 不同发酵阶段的豆酱样品中细菌16S rDNA V3区PCR-DGGE特异性条带比对结果
Table 1 Identification of the bands obtained by PCR-DGGE of bacterial 16S rDNA from soybean paste based on BLAST comparison in GenBank

根据测序结果利用Mega 5 软件构建系统发育树,结果如图所示。由图2可知,所有阶段性豆酱样品中共检测到了15 株乳酸菌种,其微生物群落可分为5 个属:四联球菌属(4 个条带)、明串珠菌属(1 个条带)、肠球菌属(3 个条带)、乳杆菌属(6 个条带)和魏斯氏菌属(1 个条带),其中乳杆菌属、四联球菌属和肠球菌属占主导地位。
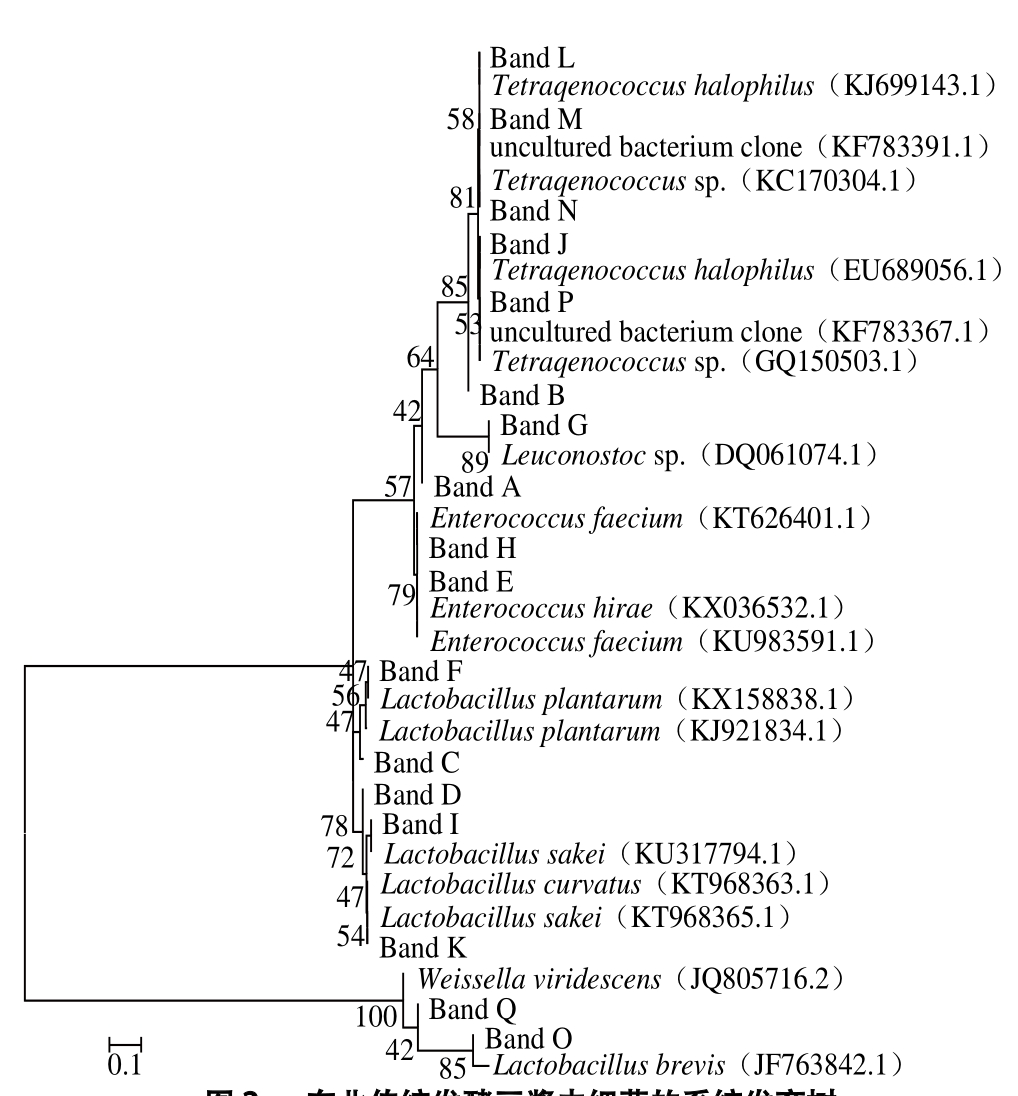
图2 东北传统发酵豆酱中细菌的系统发育树
Fig. 2 Phylogenetic tree for bacterial community in traditional fermented soybean paste
分别对四联球菌属(条带J、L、M、P)、明串珠菌属(条带G)进行检测,结果显示其分别属于嗜盐四联球菌(Tetraqenococcus halophilus)、明串珠菌(Leuconostoc sp.);肠球菌属(条带A、E、H)经检测分别属于屎肠球菌(Enterococcus faecium)、希氏肠球菌(Enterococcus hirae);乳杆菌属(条带C、D、F、I、K、O)经检测分别属于植物乳杆菌(Lactobacillus plantarum)、清酒乳杆菌(Lactobacillus sakei)、弯曲乳杆菌(Lactobacillus curvatus)和短乳杆菌(Lactobacillus brevis),魏斯氏菌属(条带Q)经检测隶属于绿色魏斯菌(Weissella viridescens)。
DGGE图谱条带的数目可以直观地反映样品中细菌菌落的遗传多样性,条带的亮度在一定程度上反映该种菌群的数目[11]。随着发酵时间的变化,图谱中的部分条带的分布发生着一定的变化,条带的数目以及条带的亮度随着发酵时间的变化在时刻发生着改变,说明各个时期的豆酱样品中细菌的数目丰度在随时上下波动。条带A(Enterococcus faecium)、G(Leuconostoc sp.)、Q(Weissella viridescens)、P(Tetraqenococcus sp.)的亮度随着发酵时间变化在变暗,说明这些微生物是启动豆酱发酵的最重要菌株,主要参与豆酱发酵初期的物质分解并在整个分解过程中起到一定的作用,其中魏斯氏菌属的菌落动态与高秀芝在传统发酵豆酱发酵过程中养分动态及细菌多样性的研究中结果一致[25];条带C(Lactobacillus plantarum)、G(Leuconostoc sp.)、H(Enterococcus faecium)、K(Lactobacillus curvatus)、L(Tetraqenococcus halophilus)、M(Tetraqenococcus sp.)、O(Lactobacillus brevis)在两家豆酱样品中均被检测到,即使来自于不同人家不同手法做制作的豆酱,乳杆菌属、肠球菌属与四联球菌属仍然稳定存在于所有豆酱样品中,说明它们为东北传统发酵豆酱在发酵过程中起中主要作用的细菌菌群;条带C、H、K与O在下酱时有一定量的存在,并且在豆酱的整个发酵阶段均很明亮、条带较宽且变化不大,说明其在豆酱发酵过程中均稳定存在,是东北传统发酵豆酱在发酵过程中的优势细菌菌种,乳杆菌属为优势菌属。乳酸菌在生长代谢过程中能与酵母菌代谢产物作用生成酯类物质,因此该菌对豆酱发酵过程的风味形成起到重要的作用。
由图1可知,不同家庭由于制作手法不同,豆酱样品中菌群也存在着一定的差异性,如条带F与Q只在Z家豆酱中被检测出;条带G的检测结果为Leuconostoc sp.,Z家豆酱中条带G的亮度较T家相比更亮,条带更宽。Kim等[26]通过16S rRNA基因的初步测序结合理化指标的测定,分析得出韩国豆酱中分离出的菌株为芽孢杆菌属(Bacillus glycinifermentans sp.);在酱的生产中,与风味有直接关系的细菌主要是乳酸菌,其中主要包括Lactobacillus plantarum、发酵乳杆菌(Lactobacillus fermenti)、Tetragenococcus halophilus等。同样,Wu Junrui等[27]在分子水平上利用DGGE技术对东北大酱中的细菌物种进行分析鉴定,得出Lactobacillus plantarum、Leuconostoc gasicomitatum、Enterococcus faecium和Tetragenococcus halophilus是豆酱中的主要细菌,本研究中的Weissella viridescens在传统自然发酵豆酱中利用传统方法和分子生态学方法均并未分离到。
2.2 OTU聚类及注释结果
OTU通常是指根据某一人为设定的序列相似度阈值,将来自一个或多个样本的序列进行归并,彼此间相似度高于该阈值的序列都将归并为一个OTU[28]。可以简化数据结构,有利于在某一确定的分类水平,对不同来源的微生物群落样本进行互相比较(表2)。
表2 豆酱样品细菌OTU数目
Table 2 Bacterial OTU number of fermented soybean paste
细菌丰度的波动趋势为0~7 d,细菌数目在增加;7~14 d,细菌数目在减少;14~21 d,细菌数目在增加,并在21 d达到了细菌数目的最大值;之后细菌数目均减少,说明在发酵前中期,豆酱的内部微生物相互作用环境与外部自然环境均有利于微生物的生长;到了发酵后期,细菌群落数目在减少,结合DGGE图谱上细菌分布动态推测,可能是Enterococcus faecium、Leuconostocsp.、Weissella viridescens和四联球菌等微生物在豆酱发酵过程中,主要参与初期的物质分解作用,其自身的代谢及其代谢产物在豆酱发酵过程中可以调整豆酱内环境,为豆酱风味物质的形成创造有利条件。其中Leuconostoc sp.能够在代谢过程中产生葡聚糖并产生风味物质;魏斯菌属于乳酸菌群,其中大多数为兼性厌氧或微需氧菌,通常分离于肉类食品中,对发酵食品风味的形成可能具有重要作用[29]。
2.3 基于UniFrac距离矩阵的UPGMA聚类分析
由于微生物极其多样,不同微生物彼此之间具有特定的系统发育关系,意味着群落中的各微生物成员(如OTU)之间存在某种内在关联。因此,在探究不同群落样本之间的关系时,需要考虑群落成员之间是否存在系统发育亲缘关系。聚类分析类群的划分显示出豆酱样品中微生物组成之间的相似性和亲缘关系,样本根据彼此之间的相似度聚类,样本间的分枝长度越短,两样本越相似。
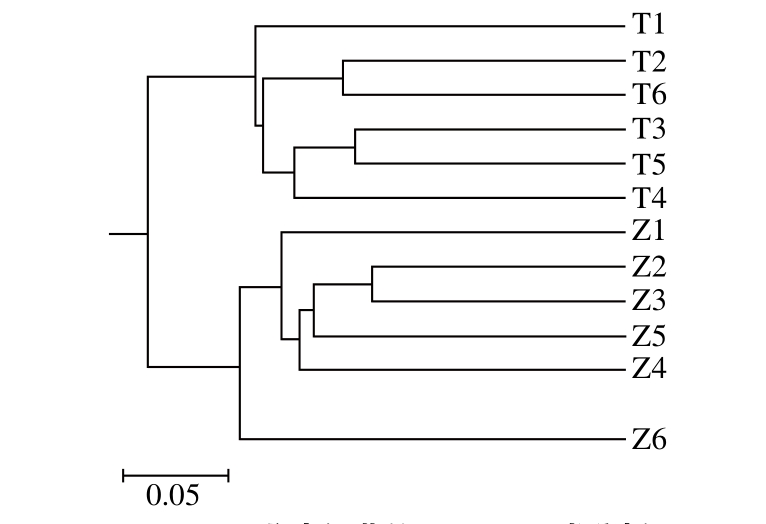
图3 豆酱中细菌的UPGMA聚类分析
Fig. 3 UPGMA cluster analysis of bacteria in soybean paste
由图3可知,两个家庭所制作的豆酱样品之间差异率较低,表明两家豆酱样品中细菌的种群结构均有较高的相似性,说明两家豆酱样品中微生物的分布特点以及具有的共性对于东北传统发酵豆酱具有一定的代表性。
2.4 豆酱样品在属水平上的物种分布
对每个豆酱样品中细菌的OTU在属水平进行分析,得到物种组成比例情况,反映样品的群落结构。
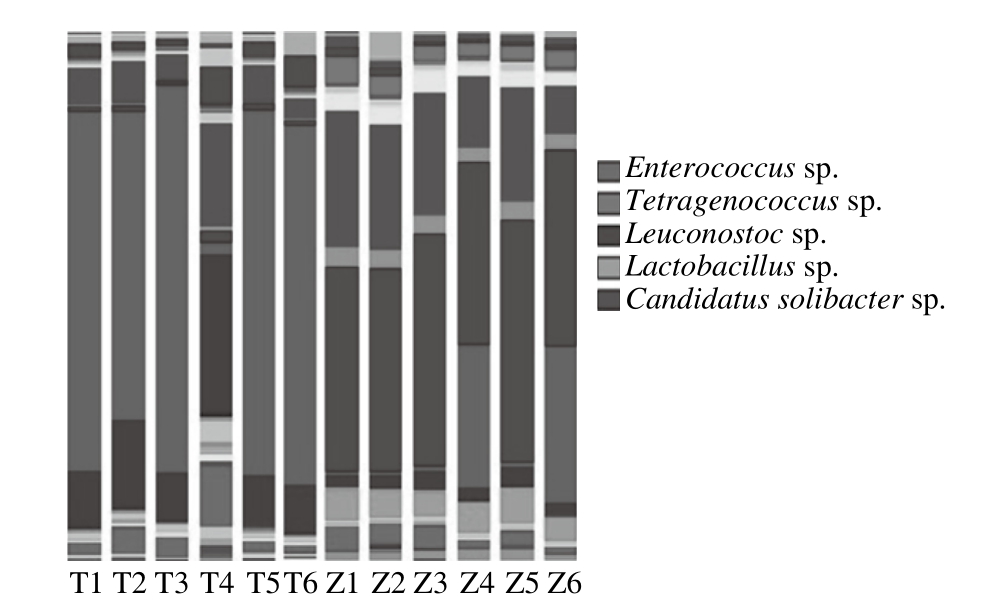
图4 豆酱样品中细菌在属水平的分布图
Fig. 4 Bacterial distribution of fermented soybean paste at the genus level
对每个豆酱样品中细菌的OTU数目在属水平上进行归类和整理,分为已知细菌属和未鉴定出的细菌属。在豆酱的不同发酵时期,样品中的细菌群落存在着明显的动态变化,其相对含量存在着一定的差别。在已知细菌属中,Leuconostoc sp.、Enterococcus sp.与Tetragenococcus sp.明显存在于所有样品中。其中,Leuconostoc sp.相对于其他菌属含量较高,相对稳定存在于所有豆酱样品中;且Tetragenococcus sp.波动性较大,在各个豆酱样品中的分布很不均匀,结合DGGE图谱可看出其对应条带在豆酱样品中的分布变化较明显,说明它易受内外环境的影响;其次数目最多的为Lactobacillus sp.,在不同发酵阶段的豆酱样品中均被检测出,且相对于其他细菌较稳定地分布于所有样品中,也验证了图2所得结论,其中Leuconostoc sp.、Enterococcus sp.、Tetragenococcus sp.和Lactobacillus sp.占主导地位。从分布图可以看出还有部分丰度相对较小的微生物群体存在于各个豆酱样品中,但由于其数目少,可能在DGGE图谱上很难明显显现出,因此只有通过DGGE技术结合高通量测序的方法才能够直观、全面、系统地对样品中的微生物群落进行探究与分析。
本实验采用PCR-DGGE分子生物学技术结合高通量测序方法对东北传统发酵豆酱在发酵过程中的细菌群落进行了多样性研究,通过DGGE图谱的分析结合高通量测序验证得出:豆酱样品不同发酵阶段的优势细菌菌属为肠球菌属(Enterococcus sp.)、四联球菌属(Tetragenococcus sp.)和乳杆菌属(Lactobacillus sp.);对豆酱样品所提供的DNA模板中高质量序列按97%的序列相似度进行归并和OTU划分,经整理与分析得出:细菌的OTU总数目为7 159 ,各个发酵阶段的细菌数目在时刻变化:在豆酱发酵前期,细菌数目上下波动,在21 d达到了细菌数目的最大值,发酵后期数目在减少,结合DGGE图谱上细菌分布动态推测可能是Enterococcus faecium、Leuconostoc sp.、Weissella viridescens等微生物在豆酱发酵过程中参与了初期的产酶降解大分子物质作用,并在前中期的大分子物质分解为小分子物质等产生风味物质的过程中起到一定的作用;对豆酱样品中细菌的OTU数目在属水平上进行归类和整理,Enterococcus sp.、Tetragenococcus sp.和Lactobacillus sp.在不同发酵阶段的豆酱样品中均被检测出,数目较其他细菌相比较多,结合DGGE结果推断其为豆酱发酵过程中的优势菌群。其中Tetragenococcus sp.波动性较大,易受内外环境的影响。
由于发酵食品自然发酵微生物区系复杂,生产周期不稳定、安全性差、品质不能保证等问题经常出现,因此研究发酵食品中微生物多样性是解决以上问题的必要途径。实验结果直观地展现出传统自然发酵豆酱中细菌的多样性及其菌相变化,因此对传统发酵豆酱样品中微生物群落的组成分布与动态规律进行研究,可以提高豆酱的品质、营养、为发掘优良的菌种发酵剂及其产业化发展提供有力的依据。
参考文献:
[1] 汤慧娟, 韩翠萍, 刘洋, 等. 传统发酵豆酱的养分变化与分析[J]. 食品与发酵工业, 2013, 39(4): 64-67.
[2] 贡汉坤. 传统豆酱自然发酵的动态分析及人工接种多菌种发酵研究[D]. 无锡: 江南大学, 2004: 1-2.
[3] 田甜. 东北传统豆酱自然发酵过程中品质特性与微生物多样性的研究[D]. 沈阳: 沈阳农业大学, 2015: 17-18.
[4] 石彦国. 大豆制品工艺学[M]. 北京: 中国轻工业出版社, 2005: 152-153.
[5] 葛菁萍, 柴洋洋, 陈丽, 等. 传统豆酱发酵过程中细菌多样性动态[J].生态学报, 2012, 32(8): 2532-2538. DOI:10.5846/stxb201103110295.
[6] 晏丽, 张银志, 王淼, 等. 自然发酵黄豆酱生产过程中理化及微生物指标的动态分析[J]. 食品与生物技术学报, 2012, 31(3): 271-275. DOI:10.3969/j.issn.1673-1689.2012.03.008.
[7] 田甜, 武俊瑞, 岳喜庆. 豆酱自然发酵过程中质地变化及相关性分析[J]. 食品与发酵工业, 2014, 40(2): 27-31. DOI:10.13995/ j.cnki.11-1802/ts.2014.02.020.
[8] 韩翠萍, 汤慧娟, 赵月, 等. 响应面优化传统发酵豆酱风味物质萃取条件的研究[J]. 大豆科技, 2015(6): 14-20. DOI:10.3969/ j.issn.1674-3547.2015.06.005.
[9] MUYZER G, de WAAL E C, UITTERLINDEN A G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA[J]. Applied and Environmental Microbiology, 1993, 59(3): 695-700.
[10] 丛敏, 李欣蔚, 武俊瑞, 等. PCR-DGGE分析东北传统发酵酸菜中乳酸菌多样性[J]. 食品科学, 2016, 37(7): 78-82. DOI:10.7506/ spkx1002-6630-201607015.
[11] 梁新乐, 朱扬玲, 蒋予箭, 等. PCR-DGGE法研究泡菜中微生物群落结构的多样性[J]. 中国食品学报, 2008, 8(3): 133-137. DOI:10.3969/ j.issn.1009-7848.2008.03.024.
[12] 张先琴. PCR-DGGE技术分析传统发酵蔬菜中微生物群落结构[D].雅安: 四川农业大学, 2012: 7-8.
[13] LIANG H P, LUO Q C, ZHANG A, et al. Comparison of bacterial community in matured and degenerated pit mud from Chinese Luzhouflavour liquor distillery in different regions[J]. Journal of the Institute of Brewing, 2016, 122(1): 48-54. DOI:10.1002/jib.296.
[14] 韩吉雨, 王海荣, 侯先志, 等. PCR-DGGE方法分析玉米及苜蓿青贮动态发酵体系中菌群多样性[J]. 安徽农业科学, 2009, 35(19): 8888-8892. DOI:10.13989/j.cnki.0517-6611.2009.19.115.
[15] 周剑忠, 董明盛. 江汉湖藏灵菇微生物种群结构的分子特性研究[J]. 中国生物学文摘, 2006, 33(4): 64-68. DOI:10.3969/ j.issn.0253-2654.2006.04.013.
[16] SHEFFIELD V C, COX D R, LERMAN L S, et al. Attachment of a 40 bp G+C rich sequence (GC clamp) to genomic DNA fragments by polymerase chain reaction results in improved detection of single-base changes[J]. PNAS, 1989, 86: 232-236.
[17] 王绍祥, 杨洲祥, 孙真, 等. 高通量测序技术在水环境微生物群落多样性中的应用[J]. 化学通报, 2014, 77(3): 196-203.
[18] 孙雯, 葛菁萍, 叶广彬, 等. 传统豆酱发酵过程中霉菌的形态学分析[J].中国农学通报 2014, 30(15): 298-304.
[19] 武俊瑞, 王晓蕊, 唐筱扬, 等. 辽宁传统发酵豆酱中乳酸菌及酵母菌分离鉴定[J]. 食品科学, 2015, 36(9): 78-83. DOI:10.7506/spkx1002-6630-201509015.
[20] 辛星, 宋刚, 周晓杭, 等. 传统发酵豆酱中乳酸菌的分离、筛选及鉴定[J]. 中国食品学报, 2014, 14(9): 202-207. DOI:10.16429/j.1009-7848.2014.09.033.
[21] 柴洋洋, 葛菁萍, 宋刚, 等. 传统发酵豆酱中酵母菌的分离、筛选及功能酵母的鉴定[J]. 中国食品学报, 2013, 13(3): 183-187. DOI:10.16429/j.1009-7848.2013.03.034.
[22] 武俊瑞. 东北传统发酵特色食品中主要微生物多样性研究[D].沈阳: 沈阳农业大学, 2013: 68-76.
[23] EDGAR R C. Search and clustering orders of magnitude faster than BLAST[J]. Bioinformatics, 2010, 26(19): 2460-2461. DOI:10.1073/ pnas.93.25.14229.
[24] LOZUPONE C, KNIGHT R. UniFrac: a new phylogenetic method for comparing microbial communities[J]. Applied and Environmental Microbiology, 2005, 71(12): 8228-8235. DOI:10.1128/ AEM.71.12.8228-8235.2005.
[25] 高秀芝, 王小芬, 李献梅, 等. 传统发酵豆酱发酵过程中养分动态及细菌多样性[J]. 微生物学通报, 2016, 35(5): 748-753. DOI:10.3969/ j.issn.0253-2654.2008.05.018.
[26] KIM S J, DUNLAP C A, KWON S W, et al. Bacillus glycinifermentans sp. nov., isolated from fermented soybean paste[J]. International Journal of Systematic and Evolutionary Microbiology, 2015, 65(10): 3586-3590. DOI:10.1099/ijsem.0.000462.
[27] WU J R, ZHANG J C, SHI P, et al. Bacterial community involved in traditional fermented soybean paste dajiang made in northeast China[J]. Annals of Microbiology, 2013, 63(4): 1417-1421. DOI:10.1007/s13213-013-0604-2.
[28] BLAXTER M, MANN J, CHAPMAN T, et al. Defining operational taxonomic units using DNA barcode data[J]. Philosophical Transactions of the Royal Society of London, 2005, 360: 1935-1943. DOI:10.1098/rstb.2005.1725.
[29] 施思, 邓宇, 李波, 等. DGGE法在盛夏习酒酒醅的微生物菌群结构解析中的应用[J]. 酿酒科技, 2010(3): 51-53.
Diversity and Dynamic Changes of the Bacterial Community during Fermentation of Soybean Paste
ZHANG Ying1, WU Rina1,2,*, SUN Huijun1,3, WU Junrui1, TAO Dongbing1, WANG Hongyu1
(1. College of Food Science, Shenyang Agricultural University, Shenyang 110866, China;
2. State Key Laboratory of Food Science and Technology, School of Food Science and Technology, Jiangnan University, Wuxi 214122, China; 3. College of Modern Agricultural Technology, Liaoning Agricultural Economic School, Jinzhou 121001, China)
Abstract:In this study, the diversity and dynamic changes of the bacterial community during the natural fermentation of soybean paste, a traditional fermented food popular in northeast China, were analyzed by using polymerase chain reactiondenaturing gradient gel electrophoresis (PCR-DGGE) technology and high-throughput sequencing. Results showed that bacterial operational taxonomy unit (OTU) values were fluctuant during the fermentation and reached a maximum on day 21, indicating the largest bacterial population. Analysis of DGGE profiles and the distribution of bacteria at the genus level suggested that Leuconostoc sp., Enterococcus sp., Tetragenococcus sp. and Lactobacillus sp. were predominant bacterial strains for different fermentation stages of soybean paste. Among these, Tetragenococcus sp. was more fluctuant and and vulnerable to both internal and external environments. Enterococcus faecium, Leuconostoc sp. and Weissella viridescens decreased with fermentation time indicating that these bacteria might be mainly involved in the production of catabolic enzymes in the early fermentation stage and played a role in the adjustment of the internal environment during the whole fermentation process.
Key words:traditional fermented soybean paste in northeast China; PCR-DGGE; sequencing for microbial diversity; dominant bacterial strains; operational taxonomy unit (OTU)
DOI:10.7506/spkx1002-6630-201714005
中图分类号:Q7
文献标志码:A
文章编号:1002-6630(2017)14-0030-06
收稿日期:2016-09-04
基金项目:国家自然科学基金面上项目(31471713;31470538);中国博士后科学基金项目(2014M560395);
辽宁省农业领域青年科技创新人才培养计划项目(2014048);
辽宁省高等学校优秀人才支持计划项目(LR2015059;LJQ2015103);江苏省博士后科研资助计划项目(1402071C)作者简介:张颖(1992—),女,硕士研究生,研究方向为食品生物技术。E-mail:794374297@qq.com
*通信作者:乌日娜(1979—),女,副教授,博士,研究方向为食品生物技术。E-mail:wrn6956@163.com