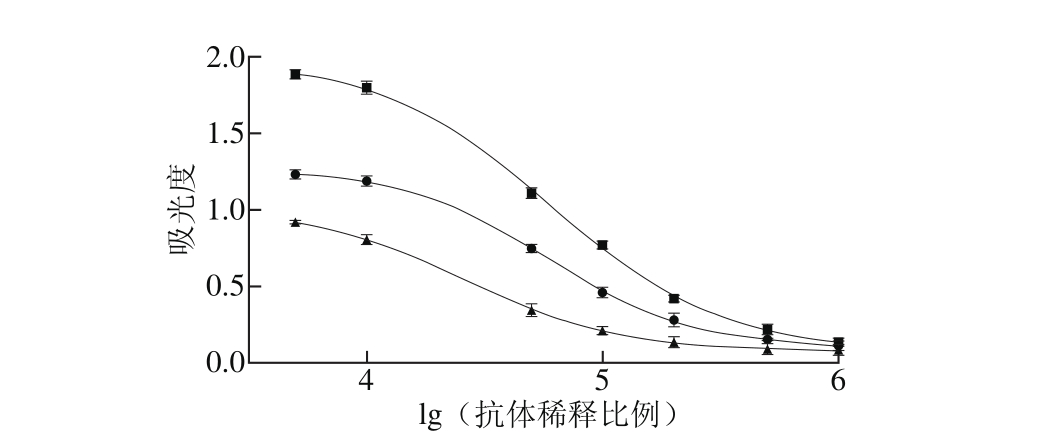
图1 EF抗体亲和常数计算
Fig. 1 Calculation of the affinity constant of anti-EF antibody
张鑫磊,隋建新,陈 静,王笑笑,林 洪,曹立民*
(中国海洋大学 食品安全实验室,山东 青岛 266003)
摘 要:目的:结合胶体金标记技术和免疫层析技术建立胶体金免疫层析毛细管检测技术,用于水产品中恩诺沙星残留的快速现场检测。方法:以间接竞争的实验原理,通过优化检测区抗原、质控区二抗、金标一抗检测浓度初步建立胶体金免疫层析毛细管检测方法,并进一步确定其检测灵敏度、特异性、重复性等及前处理的优化操作。结果:该方法检测恩诺沙星的视觉检测限为5 ng/m L,半定量检测限为1.29 ng/m L。在大菱鲆、鲅鱼、舌鳎、南美白对虾阴性样品中的平均添加回收率在78.3%~129%之间,相对标准偏差均小于10%。特异性实验表明,除对环丙沙星外,对其他相似物的特异性较好;环丙沙星的视觉检测限为10 ng/m L,半定量检测限为4.37 ng/m L,满足国标检测的限量要求。同时,针对免疫层析毛细管检测的前处理方法进行初步探究,最终确定采用常温条件下磺基水杨酸提取的前处理方法。结论:恩诺沙星胶体金免疫层析毛细管检测技术具有简单、快速、易操作、重复性好的优势,为食品药物残留检测领域提供了检测新方法。同时,选择新型试剂作为胶体金免疫层析毛细管检测技术的前处理方法,该方法可以为多种快检技术的前处理提供理论基础与技术支撑。
关键词:恩诺沙星;免疫层析技术;毛细管;胶体金
恩诺沙星(enrofloxacin,EF)是人工合成的第3代氟喹诺酮类药物,由于它具有抑菌范围广、吸收快、药物在体内存留时间长、药效好的优点,被广泛应用于水产养殖和兽医临诊中。近年来,随着食物链富集现象,EF影响人体健康而受到广泛重视与约束[1-3]。目前针对EF药物残留的检测方法有很多,包括以高效液相色谱、液相色谱与质谱联用、毛细管电泳分析法等为代表的大型仪器检测法[4],其灵敏度高,但需要使用大型仪器,检测用时较长,前处理复杂;以酶联免疫吸附法[5]、胶体金试纸条[6]为代表的传统快速检测方法,操作简便、用时较短,但具有抗基质干扰能力低、材料易受损等局限性;以荧光微球免疫法、化学发光免疫法[7-8]为代表的新型检测方法,检测灵敏度高,结果易观察,但过程较复杂,生产成本较高。因此,为了维护水产品市场的安全性和检测需求,开发一种简单、准确、可实现即时即效的检测方法,逐渐受到广泛的重视。胶体金免疫层析毛细管技术[9]是本课题组研发的一种集合胶体金标记和免疫层析的新型检测技术,该方法以玻璃毛细管为基底,背景值低,检测结果易观察;且过程简单,易操作,对检测人员的操作要求较低;对食品中硝基呋喃类代谢物[10]和“瘦肉精”[9]的初步检测应用表明,具有较高的灵敏度和重复性,可初步满足快检要求。本研究选用胶体金免疫层析毛细管技术,建立EF胶体金免疫层析毛细管检测方法,并对于基质干扰及前处理方法进行研究,进一步提高其简便性,最终利用不同水产品进行了实际应用效果验证。
1.1 材料与试剂
实验阴性样品:大菱鲆、舌鳎、南美白对虾 青岛家乐福超市;鲅鱼 青岛佳世客超市。
氯金酸、柠檬酸三钠、三乙胺 国药集团化学试剂有限公司;牛血清白蛋白(bovine serum album in,BSA)、γ-缩水甘油醚氧丙基三甲氧基硅烷(3-glycidylo xypropyltrimethoxysilane,GPTMS) 美国Sigma公司;EF、盐酸环丙沙星等标准品 中国兽医药品监察所;鼠抗EF抗体 宝安康公司;聚丙烯酰胺凝胶制备试剂盒索莱宝公司。
1.2 仪器与设备
毛细管 华西医科大学仪器厂;精密pH计 上海精密科学仪器;高速冷冻离心机 美国Sigma公司;HH.Bll.500-5电热恒温培养箱 上海跃进医疗器械厂;G4050扫描仪 中国惠普公司。
1.3 方法
1.3.1 测定原理
玻璃毛细管经强酸、强碱、有机试剂清洗处理后暴露羟基基团,再经硅烷试剂的修饰[10],可通过结合环氧基团[11]共价连接目的蛋白质,将包被原(EF-BSA)和羊抗鼠二抗分别固定在玻璃毛细管两端[12],作为检测区(TZ)和质控区(CZ)。当目标药物EF与金标一抗以一定比例混合注入检测区时,溶液中的EF与毛细管上的EFBSA同时竞争结合金标一抗,使金标一抗结合在检测区的颜色随EF含量的增大而逐渐变浅甚至消失,即检测结果判定为阳性;同样,颜色未变浅表现为酒红色的混合液,即判定结果为阴性。而质控区的二抗与金标一抗的结合显色均为酒红色,与EF含量无关,用以证明金标一抗的结合活性,且质控区显色,结果判定才有效,否则会出现假阳性、假阴性的现象,影响检测结果。
1.3.2 胶体金及金标抗体的制备
采用柠檬酸还原法[13]。通过添加不同的还原剂(柠檬酸三钠)制备不同粒径的胶体金粒子溶液。本实验在100 m L HAuCl4(1 mmol/L)在冷凝条件充分沸腾时,快速加入10 m L 38.8 mmol/L柠檬酸三钠溶液,待颜色由淡黄变为深红色停止加热,冷却后过0.22 µm水系膜,制备出13 nm胶体金溶液。
根据Slot等[14]的方法选择出最优的pH值与蛋白标记量,制备金标抗体方面略有改动。10 m L胶体金溶液中加入0.1 mol/L的K2CO3溶液,并缓慢加入适量的的鼠抗EFIgG抗体,使溶液保持约pH 8.0,缓慢均匀搅拌1 h后,加入5% BSA和1%的聚乙二醇(PEG 20000)溶液至最终体积的1/10,继续搅拌1 h后,3 500 r/m in、4 ℃离心15 m in除去暗红色沉淀,取上清液以10 000 r/m in、4 ℃离心65 m in收集沉淀,将沉淀溶于含有1% BSA、0.02% NaN3、5%蔗糖、0.1% Tween-20的1 m L Tris-HCl缓冲液(pH 8.2),重复离心收集沉淀,复溶至1 m L,置于-20 ℃备用,有效期6 个月。
1.3.3 毛细管的洗涤与修饰
对毛细管进行简单的清洗及处理后[10],将清洗干净的毛细管浸没于含有10% GPTMS和1%三乙胺的无水甲苯的修饰液中,37 ℃过夜培养后,置于大平皿中,先用无水甲苯振荡清洗3 次,每次约5 m in,再用丙酮振荡清洗3 次,每次约5 min。室温干燥保存备用,有效期4 个月[11,15]。
1.3.4 毛细管的组装与检测
分别将2 µL的抗原和二抗依次注入到毛细管两端,即为检测区和质控区,37 ℃包被反应40 m in,用含0.1%吐温-20的0.01 mo l/L,pH 7.4磷酸盐缓冲溶液(phosphate buffered solution,PBS)(即洗涤液PBST,0.01 mol/L,pH 7.4)抽洗3 次,每次用洗涤液PBST约5 m L。再将毛细管全部浸入2% BSA溶液中,37 ℃封闭2 h,用PBST洗净并室温吹干,浸没于0.01 mol/L,pH 7.4磷酸盐缓冲溶液中、4℃保存并于3个月之内使用[10]。
取3 µL金标抗体先注入检测区,待反应15 m in后,将金标抗体溶液推入质控区,常温反应15 m in后,用PBST洗净吹干,用扫描仪进行灰度扫描。
1.3.5 包被液及金标一抗工作浓度的优化
将包被原EF-BSA、羊抗鼠二抗配制成一定质量浓度梯度的稀释液,分别包被在毛细管的一端,3 组平行,检测方法同1.3.4节。以检测区与质控区显色较深的最小包被量为包被原与二抗最适的包被质量浓度。
将优化好的包被原和二抗分别固定于毛细管两端,取3 µL不同稀释比例的金标一抗分别注入检测区和质控区,方法同上。以检测区与质控区显色较深且不再增加的最大稀释比例为最适的金标抗体工作比例。
1.3.6 胶体金免疫层析毛细管检测方法的性能实验
1.3.6.1 灵敏度实验
将系列质量浓度梯度的标准EF溶液与金标一抗混合后,注入毛细管检测区,按照上述方法检测。运用相对灰度比值与EF质量浓度可形成四元拟合确定其半定量检测限,并以视觉能分析的前提下确定视觉检测限。
1.3.6.2 重复性实验
按照优化后的最佳条件组装5 批次的胶体金免疫层析毛细管,随机抽取3 根毛细管检测质量浓度3 ng/m L与12 ng/m L EF标准溶液;并随机抽取同一批次的30 根毛细管,同法检测3 ng/m L与12 ng/m L EF标准溶液,分别验证批间与批内重复性。
1.3.6.3 特异性实验
分别检测质量浓度为100、101、102、103、104、105ng/m L的EF、盐酸环丙沙星、诺氟沙星、沙拉沙星、氯霉素、土霉素等不同药物,验证EF胶体金免疫层析毛细管检测法的特异性。
1.3.6.4 胶体金免疫层析毛细管检测技术前处理的探究
在崔梦琪等[16]方法研究结果的基础上,结合磺基水杨酸与加热处理进行样品前处理研究,即取1.0 g空白样品(南美白对虾、大菱鲆、草鱼)添加2 m L 2%磺基水杨酸溶液匀浆后,分别置于室温及水浴60、70、80、90 ℃的条件下处理15 m in,4 500×g、4℃离心15 m in,取上清液用2 mol/L NaOH溶液调pH 8.0~8.5,并过0.45 µm膜制成待测溶液。通过考马斯亮蓝法测定蛋白质量浓度,将蛋白质量浓度调至1 mg/m L并使用聚丙烯酰胺凝胶制备试剂盒制备凝胶进行电泳分析,电泳操作参照陆宗超的方法[17]。进行电泳时,设置电泳初始电压为80 V,约30 m in后浓缩胶和分离胶交界处,各泳道样品保持一条直线时,将电压上升至120 V,继续1 h左右,溴酚蓝指示剂处于电泳槽底部,停止电泳,将凝胶轻轻取出,置于考马斯亮蓝染色液中,水平振荡染色2 h,再于脱色液中脱色过夜。最后,将凝胶取出并保持湿润状态,置于扫描仪中扫描,分析探究不同温度对水产品蛋白降解效果,优选出快速、高效的前处理方法。
1.3.6.5 在实际样品中的性能验证
选取大菱鲆、鲅鱼、舌鳎、南美白对虾为实际样品,经黄海水产研究所采用农业部1077号公告—1—2008《水产品中17 种磺胺类及15 种喹诺酮类药物残留量的测定 液相色谱-串联质谱法》的检测方法验证均为EF与环丙沙星阴性样品。样品处理参照崔梦琪[16]的方法并稍作改动:取(1.0±0.1)g空白样品添加EF标准溶液使其相对于水产样品的含量分别为6、30、100 µg/kg(样品),室温振荡2 h使混合物充分混匀,添加2 m L 2%磺基水杨酸溶液匀浆1~2 m in后,4 500×g、4 ℃离心15 m in,取上清液用2 mol/L NaOH溶液调pH 8.0~8.5,并过0.45 µm膜制成待测溶液,置于4 ℃避光保存,检测时与金标抗体按一定比例混合,测定其回收率与相对标准偏差,验证方法的准确性。盐酸环丙沙星样品前处理时药物加标量为16、60、160 µg/kg(样品),其他操作同EF。
2.1 鼠抗EF抗体亲和常数的计算
图1 EF抗体亲和常数计算
Fig. 1 Calculation of the affinity constant of anti-EF antibody
亲和常数主要用于描述抗体的活性,表示抗原与抗体之间特异性结合的紧密程度[18]。本实验采用常用的非竞争结合法,以抗原与抗体结合程度达到饱和程度一半即最大吸光度一半时的抗体浓度的倒数为抗体的亲和常数。通过四参数拟合标准曲线见图1,并结合Beatty推导的亲和常数公式[19],计算鼠抗EF抗体的亲和常数为1.35×107L/mol,满足检测方法建立的要求。
2.2 胶体金标记抗体复合物的制备
胶体金颗粒容易受到电解质、pH值、温度等因素的影响而发生聚沉[20],本实验通过控制胶体金溶液的K2CO3与抗体质量浓度优化制备金标抗体的pH值与抗体用量,以保持红色的最低质量浓度K2CO3与抗体质量浓度为最优[21],因此,确定了在pH 8.0、蛋白质量浓度为24 μg/m L的条件下,制备出稳定的胶体金-抗体蛋白复合物[22]。为了使胶体金标记抗体复合物维持较为稳定的溶液体系,在实际制备中选取(100+20)%的抗体标记量比例[23]即28.8 μg/m L进行标记。
2.3 胶体金免疫层析毛细管的组装
2.3.1 包被液组装浓度的优化
将不同质量浓度梯度包被抗原EF-BSA与羊抗鼠二抗包被在毛细管上,通过与抗体显色反应,选取保持稳定红色的最小抗原与二抗量为最优包被量。由图2可知,包被抗原EF-BSA质量浓度为0.075 mg/m L,羊抗鼠二抗质量浓度为0.69 mg/m L。

图2 检测区(A)与质控区(B)组装质量浓度的优化
Fig. 2 Optim ization of EF-BSA concentration in the test zone and secondary antibody concentration in the control zone
2.3.2 胶体金标记抗体检测比例的优化
按照已经确定好的抗原和二抗的包被量进行毛细管的组装,进一步确定一抗的检测比例。本实验通过1∶1~1∶6的一抗稀释比例如图3所示,选取使检测区与质控区颜色相近且肉眼易区分的稀释比例为最优,且通常情况下相对灰度值在27以上均易观察。由于胶体金免疫层析毛细管的检测灵敏度与稀释比例呈正相关,因此确定胶体金标记抗体与样品稀释液体积比为1∶3,以保证在灵敏度与肉眼易区分中都达到较好的效果。

图3 胶体金标记抗体检测浓度的优化
Fig. 3 Optim ization of the concentration of colloidal gold-labeled antibody
2.4 胶体金免疫层析毛细管检测方法的性能
2.4.1 胶体金免疫层析毛细管的灵敏度测试
根据添加不同质量浓度梯度的标准溶液进行毛细管的检测,并确定视觉检测灵敏度与半定量检测限。如图4所示,随EF标准溶液质量浓度的递增,检测区颜色由明显的红色逐渐变浅,甚至消失。根据M artina的视觉检测限定义,视觉检测限[24]是以肉眼可区分检测区颜色明显浅于阴性对照组的检测区颜色的最低EF含量表示,因此可确定胶体金免疫层析毛细管检测EF的视觉检测限为5 ng/m L。
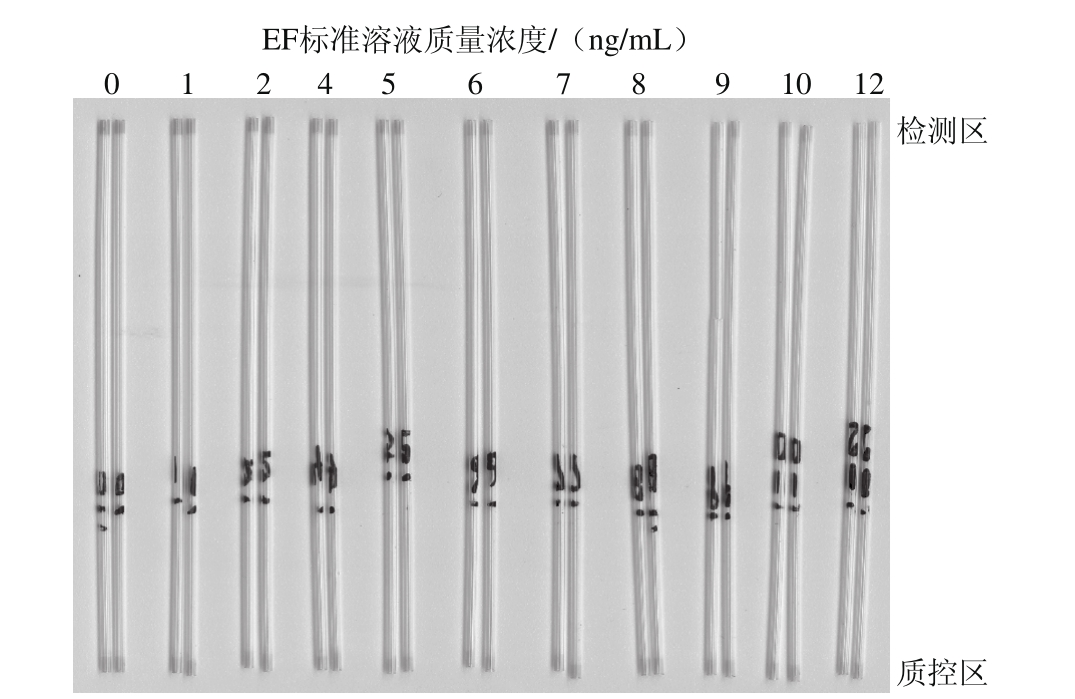
图4 EF视觉检测限显色结果
Fig. 4 Visual detection lim it for enrofloxacin
半定量检测限的确定是结合图像扫描处理技术,通过扫描仪进行灰度扫描,此时得到3 组灰度值为检测区灰度Bt、质控区灰度值Bc、背景灰度值Bb,按公式计算相对灰度值[9]:实验组检测区相对灰度值BT(BT=Bb-Bt),实验组质控区相对灰度值BC(BC=Bc-Bt),空白组的检测采用PBS缓冲液代替EF标准溶液进行测定,其空白相对灰度值B0(B0=Bb-Bt0)。通过相对灰度比值BT/B0对EF标准溶液浓度形成四参数拟合曲线如图5所示,根据得到的拟合方程计算,以3 倍空白值的标准偏差定义为EF胶体金免疫层析毛细管检测技术的半定量检测限[25]。所以根据方程计算所得的半定量检测限为1.29 ng/m L。
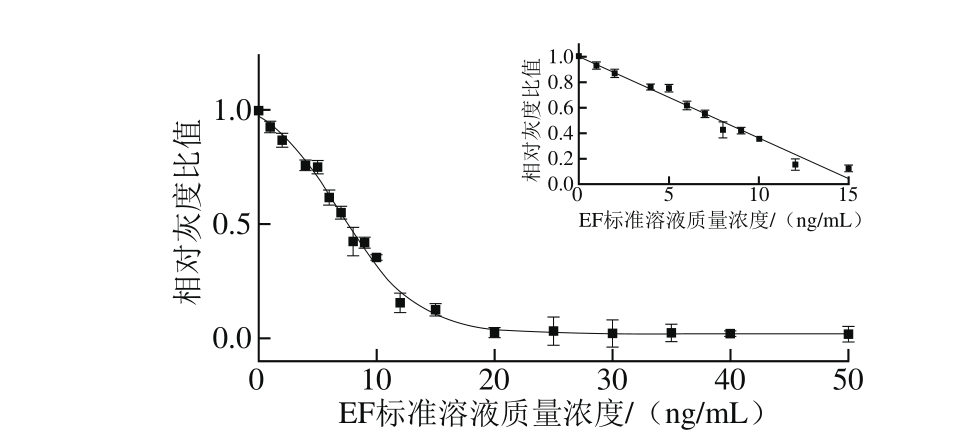
图5 胶体金免疫层析毛细管检测相对灰度比对EF质量浓度的关系曲线
Fig. 5 Calibration curve constructed by p lotting BT/B0ratio against EF concentration
2.4.2 胶体金免疫层析毛细管重复性实验结果
对于5 个批次组装的胶体金免疫层析毛细管,检测3、12 ng/m L的EF样品,得出5 批次的批间差异相对偏差为2.04%、1.72%(n=5);对于同一批次组装的20 根胶体金免疫层析毛细管检测3、12 ng/m L的样品,其批内相对标准偏差为1.72%、1.43%。说明胶体金免疫层析毛细管检测样品时显色重复性好,且高浓度使检测区颜色消失的视觉检测结果重复性也稳定,因此,胶体金免疫层析毛细管检测技术可稳定的检测EF药物残留。
2.4.3 胶体金免疫层析毛细管特异性实验结果
表1 胶体金免疫层析毛细管特异性鉴定
Table 1 Specificity of CICA for detection of enrofloxacin analogues
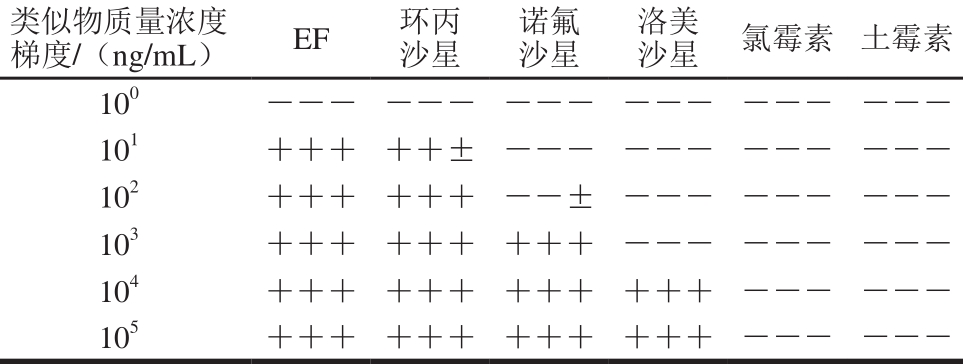
注:+.阳性,测试组检测区红色深度与阴性对照组相比显著变浅;-.阴性,测试组检测区红色深度与阴性对照组一致;±.弱阳性,测试组检测区颜色介于上述两者之间难以区分。
针对EF的胶体金免疫层析毛细管检测的特异性如表1所示,对于EF在鱼类等动物体内的代谢物环丙沙星,具有显著的交叉反应率,以同样的方法测定环丙沙星显色的检测限,并形成四参数拟合曲线如图6所示,确定环丙沙星的半定量检测限和视觉检测限分别为4.37、10 ng/m L;诺氟沙星和洛美沙星同属于氟喹诺酮类药物,结构上与EF较为类似,因此在较高质量浓度下也存在不同程度的交叉反应率[26];对于氯霉素和土霉素等族外抗生素,当质量浓度达到105ng/m L时,仍然没有出现阳性结果,说明本方法对于这些药物具有很高的特异性。
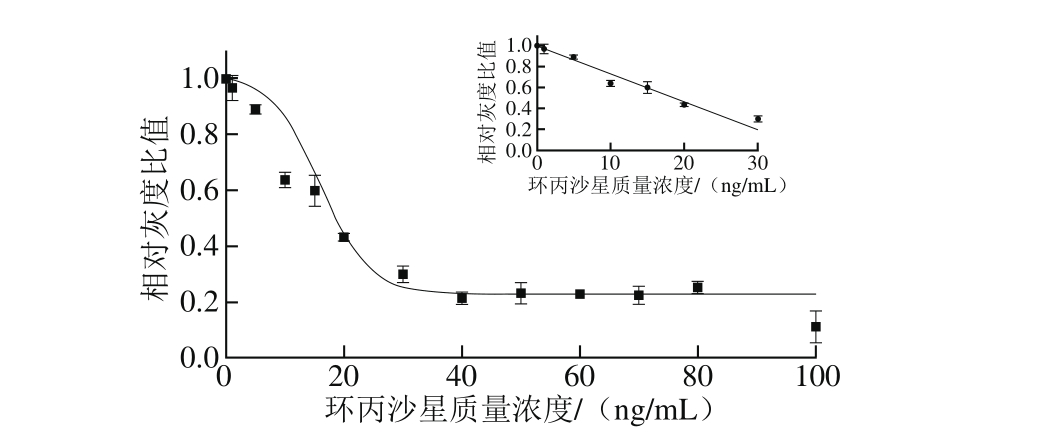
图6 胶体金免疫层析毛细管检测相对灰度比对环丙沙星质量浓度的标准曲线
Fig. 6 Calibration curve constructed by plotting BT/B0ratio against CIP concentration
据了解,各国及组织对EF的残留检测要求不同,但针对EF的检测指标,均是对EF及其代谢产物环丙沙星的共同检测结果进行限定,其中日本“肯定列表”规定EF在日本鱼贝类的限量[27]为10 µg/kg,欧盟规定其在畜禽肌肉内脏中的限量[27]为30 µg/kg,而中华人民共和国农业部公告第235号《动物性食品中兽药最高残留限量》规定EF检测限量[28]明确规定为EF与环丙沙星的检测总量不得超过100 µg/kg。本实验建立的EF胶体金免疫层析毛细管方法能够同时识别EF和环丙沙星两种药物残留,其视觉检测限分别为5 ng/m L和10 ng/m L。由此推算,本方法的检测限量是充分满足农业部235号公告中规定的限量要求,可作为EF现场检测的半定量筛检方法。
2.4.4 胶体金免疫层析毛细管检测技术前处理的探究
参考文献[29-31]发现蛋白质对检测效果影响很大,因此选择崔梦琪建立的磺基水杨酸前处理方法[16]应用到胶体金免疫层析毛细管检测方法上,对南美白对虾、大菱鲆、草鱼样品采用磺基水杨酸前处理方法结合不同温度热处理,将处理后的溶液过膜测定其蛋白含量变化,并进行聚丙烯酰胺凝胶电泳测定,如图7、8所示。

图7 基于磺基水杨酸的不同前处理方法对蛋白含量的影响
Fig. 7 Effect of different p retreatment methods w ith sulfosalicyclic acid on protein content
磺基水杨酸在室温条件下,会使3 种水产品的蛋白质量浓度明显降低;随温度升高,蛋白质量浓度的降解趋势变缓。在70 ℃水浴加热处理时,3 种水产品的蛋白质量浓度降至最低,而随温度继续升高,蛋白质量浓度均呈现波动现象。进一步结合电泳结果如图8所示,虾样品在磺基水杨酸与不同温度的处理方法上,呈现出比较稳定的规律,即随温度升高,对杂蛋白的降解效果越好;在大菱鲆、草鱼的样品处理上,温度的影响却不明显,在80、90 ℃的高温状态下,可以明显看出杂蛋白聚集、拖带、甚至重新聚合成一个新的蛋白条带。
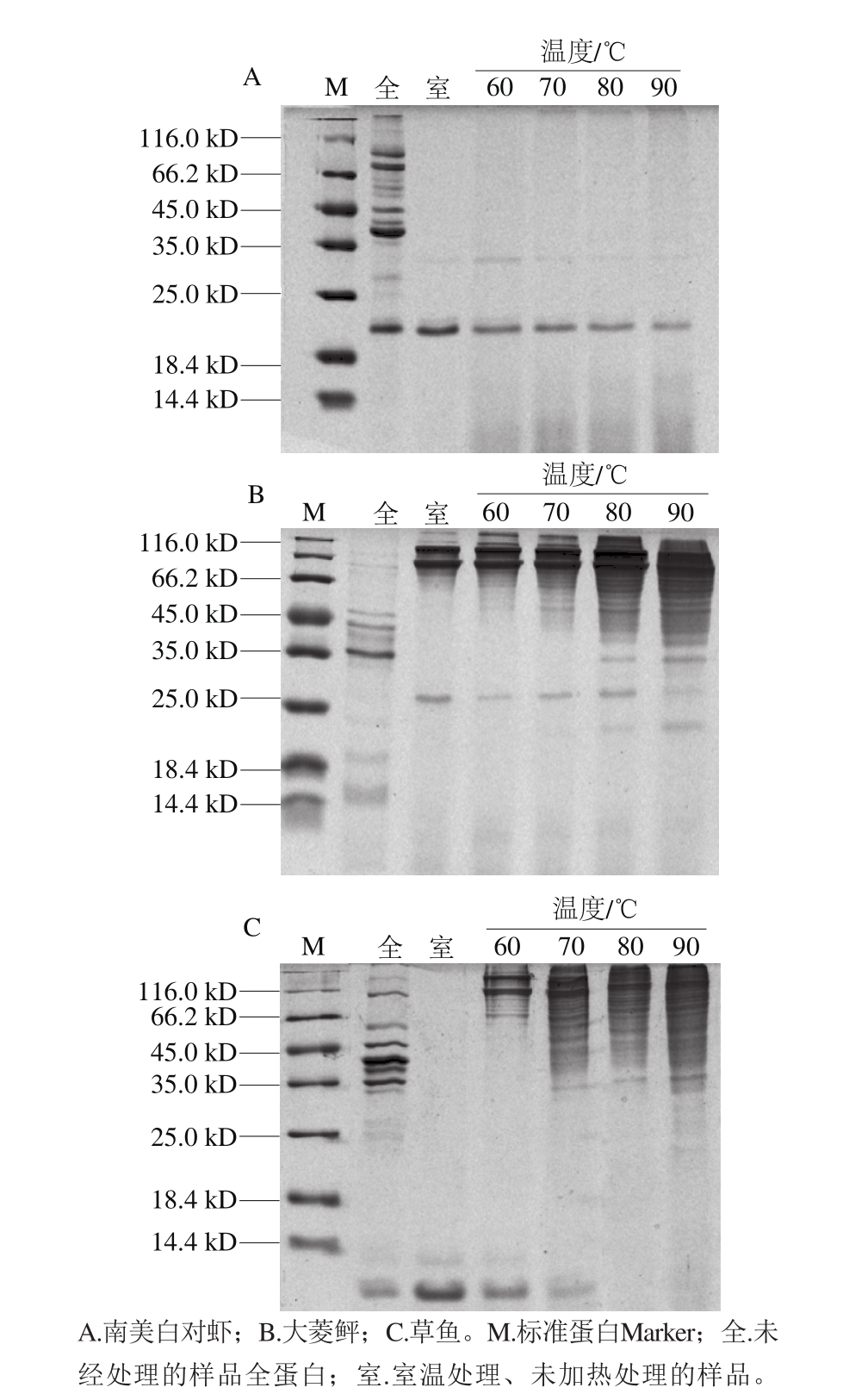
图8 基于磺基水杨酸的不同前处理方法的比较
Fig. 8 Com parison of different p retreatment methods based on sulfosalicyclic acid
结合蛋白质量浓度与种类变化规律分析原因,可能是高温与磺基水杨酸结合处理使蛋白质破坏,形成交联;随温度升高使交联的蛋白质进一步破坏、重组,进而形成蛋白条带的弥散现象。这与Petruccelli等[32]发现高温使蛋白变性进一步亚基聚合的结论相同,而且郭凤仙等[33]表示蛋白质热变性中产生的稳定聚集体,其主要原因是二硫键的形成。因此,高温结合磺基水杨酸的前处理方法,使水产品产生的新的交联蛋白质,可能对胶体金免疫层析毛细管检测造成二次干扰;同时考虑到前处理的简便程度,本实验的前处理方法采用室温条件下磺基水杨酸处理水产品,进行药物残留的检测。
2.4.5 胶体金免疫层析毛细管对实际样品的检测
在验证为阴性样品的大菱鲆、鲅鱼、舌鳎、南美白对虾中分别添加EF和盐酸环丙沙星,每个添加水平的样品分别检测3 次,随着EF添加量的增加,毛细管检测区的视觉颜色逐渐变浅,当EF和环丙沙星添加量分别达到100 µg/kg和160 µg/kg时,颜色几乎消失。通过在四参数拟合得到的EF与环丙沙星添加回收率如表2所示,平均回收率在76.84%~129%之间,相对标准偏差均小于10%,符合检测技术的要求。通过多种复杂样品的检测验证,说明EF胶体金免疫层析毛细管有很强的适用性与实用性,为胶体金免疫层析毛细管检测技术的推广提供了方向。
表2 实际样品添加回收实验结果
Table 2 Recoveries of the method for different spiked real samples

2.4.6 胶体金免疫层析毛细管检测方法与常用快速检测方法的比较
近年来,快速检测方法越来越受到广泛的重视,对检测的准确性、灵敏度、精密度、操作的简便性等综合性能的要求也愈加严格。由于本研究所用的抗体由宝安康公司提供,因此,将胶体金免疫层析毛细管检测方法与目前宝安康公司的商业化试纸条进行了主要性能的对比,同时也与部分水产品快检相关文献的研究结果进行了对比,,结果如表3所示,胶体金免疫层析毛细管检测技术呈现出相对较高的优越性,如:检测全程用时上从1~2 h明显减少到25 m in;与市场用检测试纸条检测限相同,但本实验的前处理仅约10 m in即可完成;不使用有机试剂,不需要大型仪器;检测取样量少;灵敏度也明显提高等优点。因此,基于这些优势,胶体金免疫层析毛细管检测技术是符合现代快速检测领域的需求,具有快速检测领域的相对竞争力。
表3 不同快速检测方法的检测限比较
Table 3 Comparison of detection lim its of different methods

本实验结合胶体金标记法与免疫层析法的原理,以玻璃毛细管为载体,建立了一种新型检测EF的胶体金免疫层析毛细管检测方法。使用该方法检测EF的视觉检测限为5 ng/m L、半定量检测限为1.29 ng/m L,同时对其代谢产物环丙沙星也具有较灵敏的视觉检测限10 ng/m L,初步实现多残留检测。对新建立的方法进行了重复性等性能与实际样品检测的验证,结果表明,该方法在检测不同水平的添加回收实验中,添加回收率均在76.84%~129%之间,相对标准偏差均在10 %以下,满足检测技术的基本要求,说明胶体金免疫层析毛细管技术能耐受水产品基质的复杂程度而较准确的检测水产品EF残留。
胶体金免疫层析毛细管检测技术秉承现场检测技术的目的,在操作简便性方面备受关注,因此,在课题组关于前处理方法优化探究的前期工作基础上,进一步探究了简便的前处理方法,将磺基水杨酸应用于胶体金免疫层析毛细管检测方法上,并结合热处理方法优化其操作简便性,初步探究磺基水杨酸与热处理相结合来沉降蛋白质的效果,从而验证并优选胶体金免疫层析毛细管检测的前处理方法。结果发现,磺基水杨酸与高温结合处理下的蛋白质确实可以被破坏,并随温度升高,被破坏的蛋白质进一步重组、聚合,形成新的干扰蛋白条带。而在室温(10~30 ℃)沉降蛋白效果明显,不易形成新的重组蛋白,可排除重组蛋白对免疫检测的二次干扰,同时考虑到操作、用时等方面,选择室温条件结合磺基水杨酸处理待测样品。本实验研究胶体金免疫层析毛细管检测技术前处理方法的简便性,采用磺基水杨酸作为前处理的主要试剂,与其他检测前处理方法不同,该方法提取液单一、不采用有机试剂,且操作简单、用量少,可满足市场检测领域中微量样品的检测。在行业快速简便的需求驱使下,前处理方法日趋简化、有效、准确。本实验探究的前处理方法,不仅可以实现现场检测,也为免疫检测尤其是易受蛋白质基质影响的检测,提供了新的前处理方法和研究思路。
本研究所建立的胶体金免疫层析毛细管检测技术为水产品中EF药物残留的检测技术提供了新思路、新方法,并有望将其推广利用在其他农兽药残留的检测领域。
参考文献:
[1] 宋红波, 吴光红, 沈美芳, 等. 恩诺沙星在水产品中残留的风险评估[J]. 渔业现代化, 2008, 35(5): 39-42. DOI:10.3969/j.issn.1007-9580.2008.05.010.
[2] 叶建美, 吴康, 吴洪丽, 等. 恩诺沙星抗菌效果及药代动力学研究进展[J]. 湖北农业科学, 2015, 54(23): 5813-5816. DOI:10.14088/j.cnki.issn0439-8114.2015.23.005.
[3] CINQUINA A L, ROBERTI P, GIANNETTI L, et al. Determ ination of enrofloxacin and its metabolite ciprofloxacin in goat milk by highperformance liquid chromatography w ith diode-array detection:optim ization and validation[J]. Journal of Chromatography A, 2003,987(1): 221-226. DOI:10.1016/S0021-9673(02)01800-9.
[4] 褚洪蕊, 唐景春. 农产品中农兽药残留检测技术与应用研究[J]. 农业环境科学学报, 2007, 26(B10): 656-661.
[5] 张冬冬, 苏超, 洪伟, 等. 动物源食品中氟喹诺酮类兽药残留检测的研究进展[J]. 安徽农学通报, 2016, 22(13): 38-41. DOI:10.3969/j.issn.1007-7731.2016.13.014.
[6] 潘明飞, 王俊平, 方国臻, 等. 食品中农兽药残留检测新技术研究进展[J]. 食品科学, 2014, 35(15): 277-282. DOI:10.7506/spkx1002-6630-201415056.
[7] 李怀明. 盐酸克伦特罗及恩诺沙星荧光微球免疫层析试纸条定量检测方法的建立[D]. 南昌: 南昌大学, 2012: 32-41.
[8] 龚云飞, 邹晓楠, 张露露, 等. 化学发光免疫法检测食品中恩诺沙星残留的研究[J]. 食品工业科技, 2014, 35(4): 66-69. DOI:10.13386/j.issn1002-0306.2014.04.046.
[9] QU X L, LIN H, DU S Y, et al. Development of a nano-gold capillary immunochromatographic assay for rapid and sem i-quantitative detection of clenbuterol residues[J]. Food Analytical Methods, 2016,9(9): 1-10. DOI:10.1007/s12161-016-0442-5.
[10] 杜淑媛. 水产品中危害物胶体金免疫层析毛细管检测技术的研究[D].青岛: 中国海洋大学, 2015: 25-90.
[11] 李德亮, 王军, 常志显, 等. 二氧化硅表面的GPTMS修饰[J]. 化学进展, 2008, 20(7): 1115-1121.
[12] GIOVANNOLI C, ANFOSSIA L, BAGGIANI C, et al. A novel approach for a non-competitive capillary electrophoresis immunoassay w ith laser-induced fluorescence detection for the determ ination of human serum album in[J]. Journal of Chromatography A, 2007,1155(2): 187-192. DOI:10.1016/j.chroma.2007.02.056.
[13] 杨玉新, 叶阳, 周有祥, 等. 四种化学还原法制备胶体金的比较研究[J]. 湖北农业科学, 2011, 50(3): 476-478. DOI:10.3969/j.issn.0439-8114.2011.03.013.
[14] SLOT J W, GEUZE H J. A new method of preparing gold probes for multiple-labeling cytochem istry[J]. European Journal of Cell Biology,1985, 38(1): 87-93.
[15] HONG D D, SOHN O J, LAM H T, et al. An optical pH sensor w ith extended detection range based on fluoresceinam ine covalently bound to sol-gel support[J]. M icrochem ical Journal, 2006, 84(1/2): 50-55.DOI:10.1016/j.m icroc.2006.04.013.
[16] 崔梦琪. 基于磺基水杨酸的水产品氟喹诺酮类药物酶联免疫检测前处理技术研究[D]. 青岛: 中国海洋大学, 2015: 30-43.
[17] 陆宗超. 大菱鲆过敏原小清蛋白的制备及氧化对其IgE结合能力的影响[D]. 青岛: 中国海洋大学, 2013: 15-17.
[18] 刘彦君, 谭婧, 章崇杰, 等. 应用曲线拟合方法求解单克隆抗体亲和常数中OD_(100)[J]. 西部医学, 2010, 22(4): 599-602.
[19] BEATTY J D, BEATTY B G, VLAHOS W G. M easurement o f monoclonal antibody affinity by non-com petitive enzym e immunoassay[J]. Journal of Immunological Methods, 1987, 100(1/2):173-179. DOI:10.1016/0022-1759(87)90187-6.
[20] 马爱团, 钟秀会, 史万玉, 等. 胶体金标记抗体的制备及Dot-IGSS检测IBDV方法的建立[J]. 黑龙江畜牧兽医, 2008(2): 65-67.DOI:10.3969/j.issn.1004-7034.2008.02.027.
[21] 宋宏新, 邹联柱, 李韵, 等. 纳米胶体金的制备及其抗体标记研究[J]. 食品科技, 2011, 36(3): 249-252. DOI:10.13684/j.cnki.spkj.2011.03.001.
[22] ZHAO Y L, ZHANG G P, LIU Q T, et al. Development of a lateral flow colloidal gold immunoassay strip for the rapid detection of enrofl oxacin residues[J]. Journal of Agricultural & Food Chem istry,2008, 56(24): 12138-12142. DOI:10.1021/jf802648z.
[23] 刘珊娜, 葛怀娜, 孟繁桐, 等. 李斯特菌免疫检测用胶体金的制备及抗体标记[J]. 食品科技, 2016, 41(1): 321-324. DOI:10.13684/j.cnki.spkj.2016.01.063.
[24] BLAZKOVA M, M ICKOVAHOLUBOVA B, RAUCH P, et al.Immunochromatographic colloidal carbon-based assay for detection of methiocarb in surface water[J]. Biosensors & Bioelectronics, 2009,25(4): 753-758. DOI:10.1016/j.bios.2009.08.023.
[25] 吕涛, 冯奇, 史利涛, 等. 分析方法检出限的确定[J]. 漯河职业技术学院学报, 2007, 6(4): 199-200.
[26] 姜金庆, 杨雪峰, 王自良, 等. 氟喹诺酮类药物多残留间接竞争ELISA检测方法的建立[J]. 中国预防兽医学报, 2011, 33(11): 887-892.
[27] 王晓艳, 林纪昀, 杨利, 等. 两株高特异性抗恩诺沙星单克隆抗体的研制[J]. 食品安全质量检测学报, 2010, 27(1): 5-10.
[28] 农业部兽医局. 动物性食品中兽药最高残留限量: 中华人民共和国农业部公告第235号[EB/OL]. (2002-12-24). http://www.moa.gov.cn/zw llm/tzgg/gg/200302/t20030226_59300.htm.
[29] 王秀丹. 鱼类主要基质组分对药物残留酶联免疫吸附检测的影响[D].青岛: 中国海洋大学, 2014: 14-31.
[30] 李桂芝, 高福凯, 张兴梅, 等. 柠檬酸水溶液生物亲和萃取-高效液相色谱法检测鱼肉样品中喹诺酮类药物残留[J]. 分析化学, 2013,41(10): 1592-1596. DOI:10.3724/SP.J.1096.2013.30385.
[31] 徐锐. 海产品中恩诺沙星残留的免疫胶体金层析现场快速检测技术[D]. 青岛: 中国海洋大学, 2013: 12-17.
[32] PETRUCCELLI S, ANON M C. Thermal aggregation of soy protein isolates[J]. Journal of Agricultural and Food Chem istry, 1995, 43(12):3035-3041. DOI:10.1021/jf00060a009.
[33] 郭凤仙. 热处理对大豆分离蛋白结构及功能特性的影响[D]. 无锡:江南大学, 2009: 8-28.
[34] 梅彬. 水产品中恩诺沙星胶体金快速检测试纸条的研制[D]. 厦门:集美大学, 2015: 12-41.
[35] 徐锐. 海产品中恩诺沙星残留的免疫胶体金层析现场快速检测技术[D]. 青岛: 中国海洋大学, 2013: 28-37.
[36] 王宵雪. 恩诺沙星免疫胶体金快速检测试纸条的研制[D]. 天津:天津科技大学, 2012: 34-45.
[37] 樊晓博, 谢兰心. 酶标抗原直接竞争ELISA检测食品中氟喹诺酮类药物多残留[J]. 食品科学, 2015, 36(24): 265-269. DOI:10.7506/spkx1002-6630-201524049.
Development of a Nano-Gold Capillary Immunochromatographic Assay and Comparison of Different Sample Pretreatments for Detection of Enrofloxacin in Aquatic Products
ZHANG Xinlei, SUI Jianxin, CHEN Jing, WANG Xiaoxiao, LIN Hong, CAO Lim in*
(Food Safety Laboratory, Ocean University of China, Qingdao 266003, China)
Abstract:Objective: To establish a nano-gold capillary immunochromatographic assay (CICA) based on a combination of colloidal-gold labeling and Immunochromatography for the rapid field detection of enrofloxacin (EF) residues in aquatic products. Methods: Based on the principle of indirect competitive immunoreaction, the CICA method was developed by optim izing antigen concentration in the test area, secondary antibody in the control area and gold-labeled antibody standard concentration. Then the sensitivity, specificity and repeatability of this method and several sample pretreatments were evaluated. Results: The visual and sem i-quantitative detection lim its for EF were estimated to be 5 and 1.29 ng/m L,respectively. The recoveries of EF in spiked negative samples of turbot, Spanish mackerel, tonguefishes, and Pacific while shrimp (P. vannamei) were 78.3%–129% and The intra- and inter-assay precision expressed as relative standard deviation(RSD) was less than 10%. Specificity analysis indicated that this method had strong specificity to all analogues except ciprofloxacin (CIP), for which the visual and sem i-quantitative detection lim it were 10 and 4.37 ng/m L, respectively and met the requirement of the national standard of China. Sulfonic acid extraction under normal temperature conditions was chosen as the optimal pretreatment method for CICA. Conclusion: CICA was a simple, rapid, easy-to-operate and reproducible method and could provide a new approach for the detection of antibiotic residues in aquatic products. Meanwhile, the pretreatment method for CICA chosen in this study can provide a theoretical basis and technical support for other rapid detection methods.
Key words:enrofloxacin; immunochromatography; capillary; colloidal gold
DOI:10.7506/spkx1002-6630-201722048
中图分类号:TS254.7
文献标志码:A
文章编号:1002-6630(2017)22-0322-08
收稿日期:2016-11-14
作者简介:张鑫磊(1990—),女,硕士研究生,研究方向为食品安全检测。E-mail:zhangxinlei.hao@163.com
*通信作者:曹立民(1972—),男,教授,博士,研究方向为食品安全检测。E-mail:caolim in@ouc.edu.cn
引文格式:
张鑫磊, 隋建新, 陈静, 等. 水产品中恩诺沙星胶体金免疫层析毛细管检测及其前处理[J]. 食品科学, 2017, 38(22):322-329. DOI:10.7506/spkx1002-6630-201722048. http://www.spkx.net.cn
ZHANG Xinlei, SUI Jianxin, CHEN Jing, et al. Development of a nano-gold capillary immunochromatographic assay and comparison of different sample pretreatments for detection of enrofloxacin in aquatic products[J]. Food Science, 2017,38(22): 322-329. (in Chinese w ith English abstract)
DOI:10.7506/spkx1002-6630-201722048. http://www.spkx.net.cn