
图1 SEM蛋白编码基因和氨基酸序列的比对
Fig. 1 Analysis of the full-length DNA and amino acids sequences of SEM
刘 骥,杨 帆,田万帆,龙 虎,孙思雨,周玉珊,赵燕英,唐俊妮*
(西南民族大学生命科学与技术学院,四川 成都 610041)
摘 要:葡萄球菌肠毒素M是由金黄色葡萄球菌νSa基因岛编码的分泌型超抗原。本研究将截去N端信号肽的金黄色葡萄球菌M型肠毒素(staphylococcal enterotoxin M,SEM)蛋白编码基因亚克隆至原核表达载体pET-28a(+),构建重组表达质粒pET-28a(+)-ΔNspsem;随后转化感受态E. coli Rosetta(DE3)并探讨融合蛋白最佳表达条件;利用Ni2+-Sepharose 4 Fast Flow亲合层析纯化出His-ΔNspSEM融合蛋白;经质谱鉴定,纯化的融合蛋白对SEM的氨基酸覆盖率达96.3%;凝血酶切除6×His标签肽后,圆二色谱分析表明ΔNspSEM重组蛋白富含β-折叠(35%)和β-转角(21%)以及较少α-螺旋(16%)等二级结构;荧光发射谱揭示ΔNspSEM在278 nm和295 nm波长处激发时具有相同的Trp发射峰(341 nm);十二烷基硫酸钠-聚丙烯酰氨凝胶电泳分析表明ΔNspSEM重组蛋白表现出较高的热稳定性。研究结果表明重组蛋白SEM表达成功,纯化的ΔNspSEM重组蛋白溶液构象紧密并具有接近天然状态的结构,这为深入研究SEM蛋白的结构与功能提供理论支持。
关键词:金黄色葡萄球菌M型肠毒素;原核表达;纯化;质谱;圆二色谱;荧光发射谱;热稳定性
金黄色葡萄球菌(Staphylococcus aureus)作为一个重要的食源性病原菌,其抗生素抗性获得[1-2]、生物被膜形成[3]和致热性毒素超抗原的胞外分泌[4]是逃避药物与机体免疫系统攻击的有效手段。其中,致热性毒素超抗原是一类特殊的非糖基化、低分子质量、在多肽链N-末端具有跨膜分泌型信号肽的细胞外毒素蛋白。当其在信号肽引导分泌到胞外,其N-末端信号肽被切除,形成分子质量范围为19~30 kD的成熟肽段[5],该家族的毒素具有过度活化免疫系统,诱导免疫细胞分泌大量细胞因子,导致体液中大量产生多种促炎细胞因子的异常免疫应答,从而引发全身炎症反应综合征,严重者可导致多器官功能障碍综合征[6]。金黄色葡萄球菌分泌的肠毒素(staphylococcal enterotoxins,SEs)和类肠毒素(staphylococcal-like enterotoxins,SEls)归属于上述的致热性毒素超抗原家族。同时,SEs/SEls也是造成世界范围内葡萄球菌食物中毒的重要毒力因子[7-8]。
金黄色葡萄球菌M型肠毒素(staphylococcal enterotoxin M,SEM)归属于V型超抗原[9-11],与其他各型肠毒素不同的是,M型超抗原具有一个附加的15 个氨基酸残基形成的插入序列,该插入序列位于第3个α-螺旋和第8个β-折叠股之间,被命名为α3-β8环,这个环可能在与T淋巴细胞表面的Vβ-TCR相互作用时发挥着关键作用[12]。由于该型超抗原缺乏催吐活性所要求的胱氨酸环结构,金黄色葡萄球菌超抗原国际命名委员会(International Nomenclature Committee for Staphylococcal Superantigens,INCSS)最初把SEM归类于SEls中,记为SElM[13]。最近,Omoe等[14]通过猕猴喂饲实验证实SEM具有催吐活性,但相对于传统的肠毒素活性弱,因此根据INCSS命名规则,建议将SElM称为SEM更为恰当。SEs具有显著的热稳定性、蛋白酶降解稳定性、酸稳定性和干燥稳定性等特点,这种超强的环境稳定性和生物学活性,使该家族蛋白中某些成员虽经极端条件的食品加工处理步骤,仍保持一定的活性[14-15]。到目前为止,针对SEM的研究不多,主要体现在毒素功能[16-17]和快速检测[18]上。特别是关于SEM高产原核表达体系建立、表达条件优化、SEM溶液构象及维持等相关研究报道较少。
鉴于SEs引起的食物中毒普遍性和高发性,为了进一步理解新型肠毒素结构与功能关系,探讨SEs高稳定性原因,本研究通过优化SEM原核表达体系获得足量融合蛋白,进而利用圆二色谱和荧光光谱对SEM溶液构象及维持进行研究,旨在揭示SEM溶液构象及其维持的分子基础,为探索破坏SEM结构稳定性的方法和建立更加高效合理的食品消毒工艺提供依据。
1.1 材料与试剂
保存于本实验室临床分离的金黄色葡萄球菌SA003;感受态大肠杆菌(E. coli)菌株DH5α、BL21(DE3)、BL21(DE3)pLysS和Rosetta(DE3)以及高保真DNA聚合酶(Pfu DNA Polymerase) 天根生化科技(北京)有限公司;原核表达质粒pET-28a(+) 美国Novagen公司;卡那霉素、氯霉素、牛血清白蛋白组分V(bovine albumin V,BSA) 美国Sigma公司;镍亲和层析介质(Ni2+-NTA-Sepharose) 美国GE Healthcare公司;聚合酶链式反应(polymerase chain reaction,PCR)扩增试剂、核酸分子质量标准、限制性内切酶NdeⅠ和XhoⅠ 宝生物工程(大连)公司。
1.2 仪器与设备
HZQ-F160全温振荡培养箱 江苏太仓培英实验设备有限公司;5804R型冷冻离心机 德国Eppendorf公司;WD800B型微波炉 中国格兰仕集团;TSNENEN031445型PCR仪、伯乐Mini-PROTEAN Tetra Cell电泳仪 美国Bio-Rad公司;DYY-6C型核酸电泳仪北京六一仪器厂;JY92-IIN型超声破碎仪 宁波新芝生物科技股份有限公司;F-7000荧光分光光度计 日本Hitachi公司;Model 400圆二色谱仪 美国Aviv公司。
1.3 方法
1.3.1 引物合成与设计
根据GenBank中报道的S. aureus 04-02981的sem基因序列(GenBank accession No. CP001844.1)和S. aureus strain H4的selm基因序列(登录号:KT853047.1),利用SignalP 4.1 Server在线服务器预测信号肽序列。在预测的信号肽剪切位点,利用Premier 6.0进行引物设计,克隆不含信号肽的sem基因序列。不含信号肽的sem基因5’端引物序列:5’-CGTACGCATATGGATGTCGGAGTTTTGAA TCTT-3’(下划线示出NdeⅠ酶切位点),3’端引物序列为:5’-CCGCTCGAGTTAACTTTCGTCCTTATAAGATA TT-3’(下划线示出XhoⅠ酶切位点,加粗斜体为插入的终止密码子)。
1.3.2 ΔNspSEM蛋白原核表达载体构建
采用本实验室建立的微波加热法提取金黄色葡萄球菌基因组DNA,以提取的1 ng SA003菌株的基因组DNA为模板,利用1.3.1节设计的引物进行PCR扩增。扩增体系如下:10×PCR Buffer 5 μL,dNTP(2.5 mmol/L)4 μL,Mg2+(25 mmol/L)5 μL,5ʹ-引物(20 μmol/L)2.5 μL,3ʹ-引物(20 μmol/L)2.5 μL,金黄色葡萄球菌基因组DNA模板1 μL,Pfu DNA Polymerase 1 μL,ddH2O 34 μL,反应体系共计50 μL。PCR参数为:94 ℃60 s;66 ℃ 60 s;72 ℃ 150 s,38 轮循环;第1次循环前先经95 ℃变性4 min,最后1次循环后于72 ℃延伸8 min。扩增所得片段经琼脂糖凝胶电泳检测及PCR产物纯化试剂盒纯化后,与质粒载体pMD18-T进行T-A连接,转化至大肠杆菌DH5α,同时将空质粒pET-28a(+)转化至大肠杆菌DH5α。分别将插入ΔNspsem基因的pMD18-T质粒与空载质粒pET-28a(+)用NdeⅠ/XhoⅠ双酶切(注:ΔN表示N端缺失,sp是信号肽signal peptide的缩写),将上述双酶切获得的基因片段用T4 DNA连接酶连接,使得目的基因定向克隆至表达载体。将插入ΔNspsem基因的质粒命名为pET-28a(+)-ΔNspsem,转化至大肠杆菌DH5α扩增,将提取到的质粒pET-28a(+)-ΔNspsem进行NdeⅠ/XhoⅠ双酶切鉴定。同时将提取到的质粒送成都擎科生物技术有限公司测序。
1.3.3 重组质粒在大肠杆菌中表达及表达条件优化
将经过测序验证的重组质粒pET-28a(+)-ΔNspsem分别转化感受态大肠杆菌BL21(DE3)、BL21(DE3)pLysS和Rosetta(DE3),在相应的抗生素抗性平板上随机挑取含有重组质粒的单克隆,分别过夜培养后,按1∶50的比例扩大至含相应抗生素抗性的LB培养基中,37 ℃振荡培养3 h,然后加入适当浓度诱导剂异丙基-β-D-硫代半乳糖苷(isopropyl β-D-1-thiogalactopyranoside,IPTG)后,继续恒温振荡培养一定时间,离心收集菌体,十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gelelectrophoresis,SDSPAGE)检测蛋白表达情况。选择高表达菌株,分别进行诱导时间梯度(37 ℃,0.5 mmol/L IPTG分别诱导4、8、16 h),诱导用IPTG浓度梯度(37 ℃,8 h,IPTG浓度分别为0.2、0.4、0.6、0.8、1.0 mmol/L)和诱导温度(8 h,IPTG浓度0.5 mmol/L,诱导温度分别为17、27、37 ℃)优化,确定最佳诱导条件。
1.3.4 His-ΔNspSEM蛋白分离纯化及质谱鉴定
根据1.3.3节确定的最优表达菌株为出发菌种接种于5 mL LB液体培养基中,37 ℃过夜振荡培养。次日将种子液转接到500 mL LB中按照1.3.3节确定的最佳表达条件扩大培养,监测菌液A600nm至0.6~0.7时,进行IPTG诱导表达。离心收集菌体,菌体经PBS缓冲液洗涤后重悬浮于20 mL Buffer A缓冲液(50 mmol/L Tris-HCl,500 mmol/L NaCl,pH 8.0)中,冰浴超声破壁,离心收集上清液。将制备的His-ΔNspSEM蛋白粗提液上样于事先用含Buffer A缓冲液平衡好镍亲合层析柱。先以Buffer A缓冲液和含50 mmol/L咪唑的Buffer A缓冲液除去大部分杂蛋白,再以含500 mmol/L咪唑的Buffer A洗脱目的蛋白,SDS-PAGE分析纯度。将纯化的His-ΔNspSEM进行SDS-PAGE,考马斯亮蓝染色直径3 mm以上,切下染色后的条带,送北京华大蛋白质研发中心有限公司进行质谱鉴定。
1.3.5 凝血酶去除6×His标签肽及ΔNspSEM重组蛋白纯化
将按1.3.4节中纯化的His-ΔNspSEM融合蛋白用截留分子质量10 kD、容量15 mL的millipore超滤管脱盐至6 mmol/L Britton-Robinson缓冲液(pH 7.0)中,作为凝血酶的酶切底物,配制为1 IU/μL Thrombin储液,按10 IU Thrombin/mg目标蛋白的酶与底物比例,恒温20 ℃分别进行0、0.5、1、2 h酶切。按上述方案酶切后的样品经镍亲和层析柱吸附6×His标签肽,收集穿透液再次超滤浓缩至6 mmol/L pH 7.0 Britton-Robinson缓冲液,调节超滤后ΔNspSEM融合蛋白的280 nm波长处吸光度使之与超滤前样品一致,SDS-PAGE验证酶切效率与纯化所得ΔNspSEM重组蛋白纯度。
1.3.6 圆二色谱测定
将1.3.5节纯化的ΔNspSEM重组蛋白质量浓度调节为0.089 5 mg/mL,在Model 400型圆二色谱仪上测定远紫外圆二色谱的扫描范围为190~260 nm。激发光和发射光狭缝均设为1 nm,扫描速度设为中速,光谱校正设为开启以消除光栅和检测器响应的波长依赖性。每个样品扫描3 次取平均值,3 次扫描差别较大时再扫描一次,测试在25 ℃条件下进行。采用DichroWeb在线分析软件,选择CDSSTR拟合方案,采用圆二色谱参考数据集SMP180,190~240 nm波长范围内对扫描数据进行拟合,计算溶液中ΔNspSEM蛋白二级结构含量[19]。
1.3.7 荧光光谱分析
将1.3.5节纯化的ΔNspSEM重组蛋白质量浓度调节为25 μg/mL,使用F-7000荧光光谱仪进行测定,激发波长分别为278 nm和295 nm,发射谱波长扫描范围为300~400 nm,扫描速率为1200 nm/min,所有扫描激发光和发射光狭缝均设为5 nm,PMT电压均设为700 V,光谱校正均设为开启以消除光栅和检测器响应的波长依赖性。每个样品扫描3 次取平均值,3 次扫描差别较大时再扫描一次,测试在25 ℃进行。
1.3.8 ΔNspSEM重组蛋白热稳定性分析
分别将去除6×His标签肽的ΔNspSEM重组蛋白、BSA和商品化高保真DNA聚合酶(Pfu DNA Polymerase)进行100 ℃热处理1 h,间隔15 min采样,将离心所得上清液和沉淀分别进行SDS-PAGE分析。
2.1 重组表达质粒pET-28a(+)-ΔNspsem的基因测序鉴定
将重组表达质粒ΔNspsem基因测序,得到长度为651 bp的片段,利用NCBI BLAST多序列比对发现克隆到的ΔNspsem基因序列与来源于S. aureus strain H4的SElM蛋白基因序列(登录号:KT853047.1)的第67~717位碱基序列具有100%的相似度。如图1A所示,利用SignalP 4.1 Server在线服务器预测,被截去的第1~66位碱基为SEM编码信号肽的碱基序列。图1B表明,含组氨酸标签并截去N端信号肽的SEM融合蛋白(His-ΔNspSEM)表达载体被成功构建。
图1 SEM蛋白编码基因和氨基酸序列的比对
Fig. 1 Analysis of the full-length DNA and amino acids sequences of SEM
2.2 ΔNspsem基因在大肠杆菌中的表达条件优化及纯化
2.2.1 编码ΔNspSEM蛋白的基因在不同大肠杆菌中诱导表达及表达条件优化
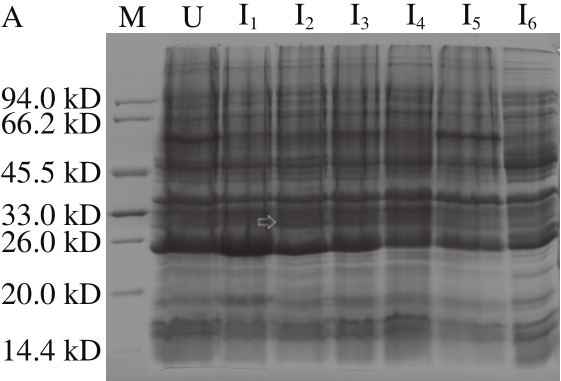
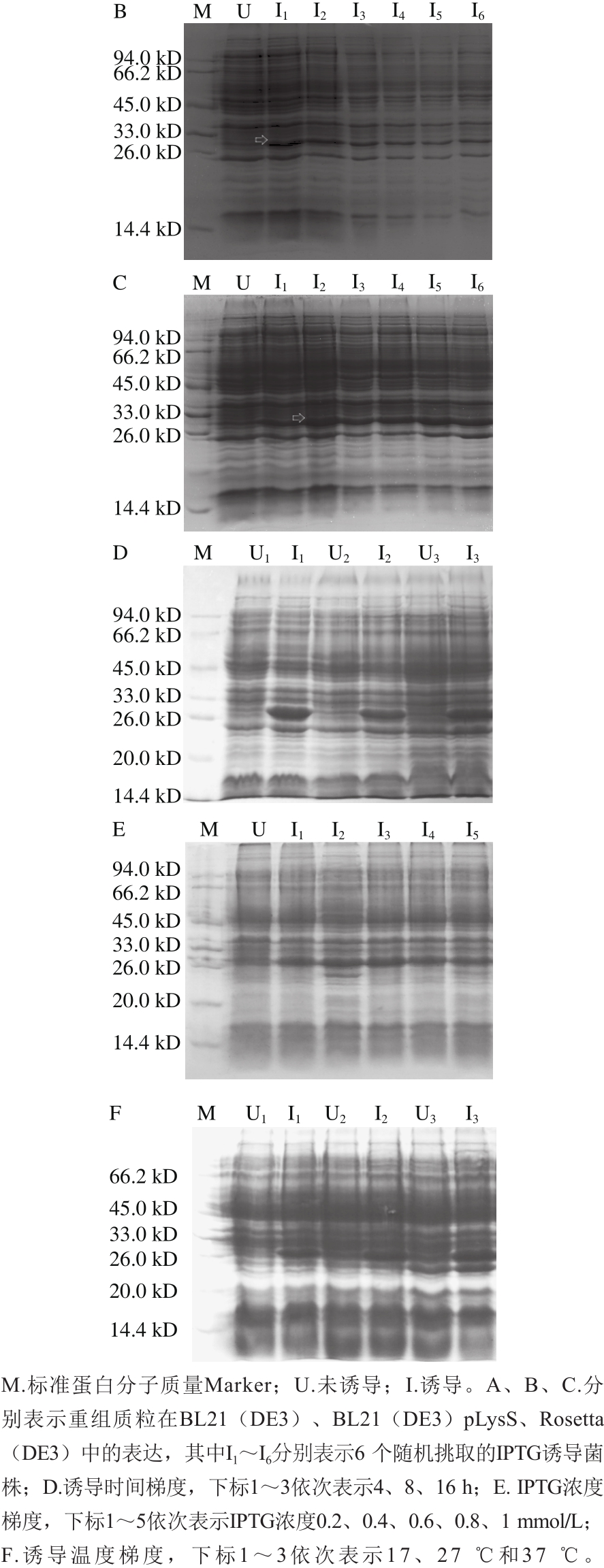
图2 编码ΔNspSEM蛋白的基因在不同大肠杆菌中诱导表达及表达条件优化
Fig. 2 Influence of expression vector and induction conditions on expression efficiency of ΔNspsem gene
编码ΔNspSEM蛋白的基因在不同大肠杆菌中诱导表达及表达条件优化时,如图2A所示,ΔNspSEM蛋白在BL21(DE3)菌株无明显表达;图2B表明在BL21(DE3)pLysS菌株中融合蛋白呈现较低的表达量;图2C显示ΔNspSEM蛋白在Rosetta(DE3)菌株中表达量普遍较高。因此,选取表达量最高的Rosetta(DE3)菌株为后续表达条件优化实验的菌种。由图2C可见,6 个重组子均能表达分子质量约为30 kD的蛋白带,与理论计算的His-ΔNspSEM蛋白分子质量接近。
进一步探讨融合蛋白的最佳表达条件,如图2D所示,诱导时间超过4 h融合蛋白表达量下降;图2E显示,融合蛋白表达量在IPTG浓度0.4~0.6 mmol/L时最高;图2F表明,37 ℃时诱导融合蛋白产量最高。确定最优表达条件为:4 h、IPTG 0.5 mmol/L、诱导温度37 ℃。
2.2.2 最优条件下His-ΔNspSEM蛋白的表达与纯化及6×His标签肽的凝血酶切除
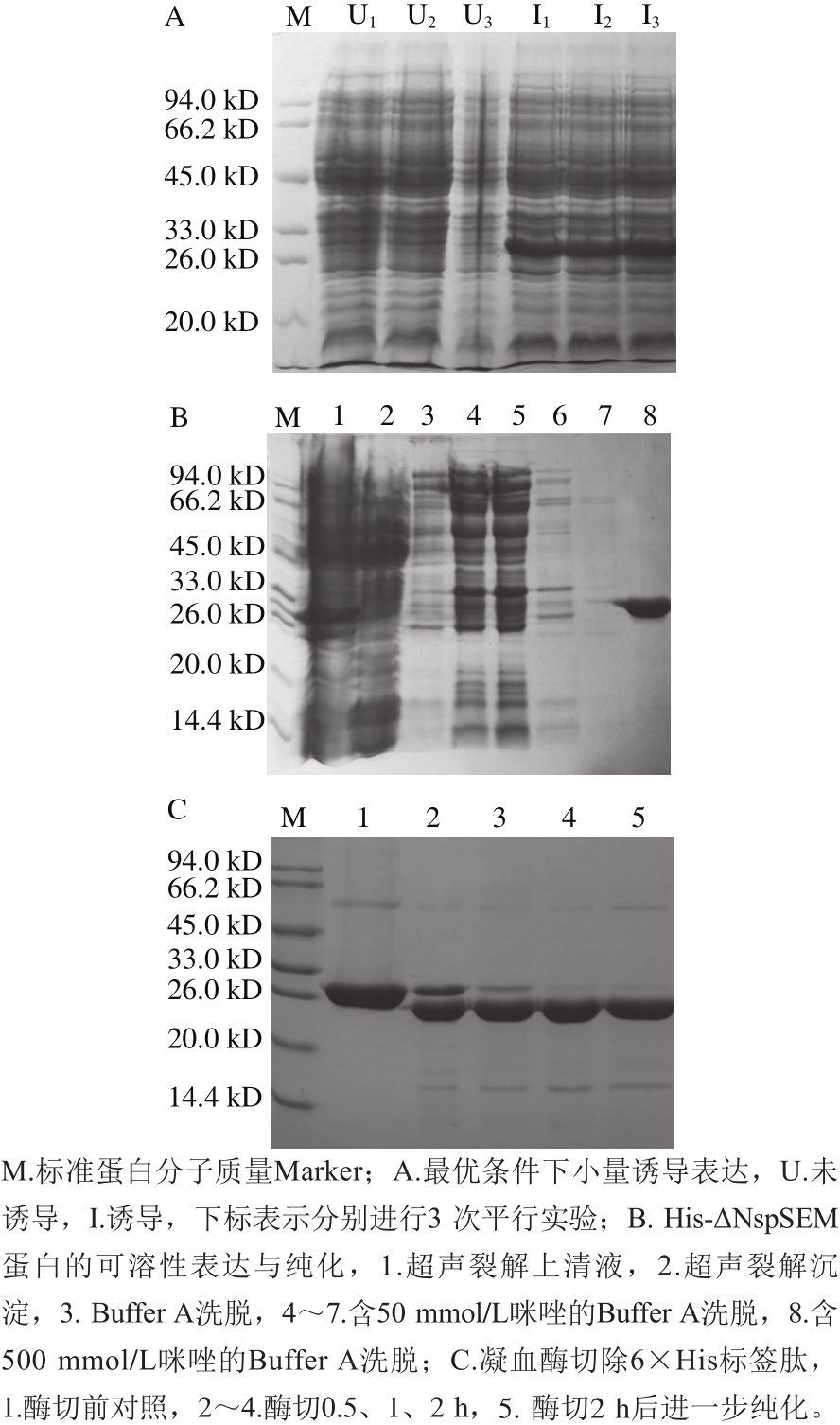
图3 最优条件下His-ΔNspSEM蛋白的可溶性表达验证与纯化
Fig. 3 Soluble expression and purification of His-ΔNspSEM protein under the optimal conditions
如图3A所示,利用2.2.1节确定的最优表达条件对Rosetta(DE3)宿主菌2号单克隆进行小量诱导表达验证,3 次平行实验均获得了His-ΔNspSEM蛋白高产量表达。将1 mL菌液离心收集菌体经过Buffer A缓冲液重悬后超声破碎,裂解液离心所得上清液(图3B泳道1)和沉淀(图3B泳道2)分别上样于SDS-PAGE,结果表明融合蛋白为可溶性表达。将扩大培养所得融合蛋白超声破碎粗提液进行镍亲合层析纯化,得纯度大于98%的His-ΔNspSEM融合蛋白。经软件分析目的蛋白分子质量约30 kD,与预期相符合(图3B泳道8)。按照1.3.5节方案凝血酶切割6×His标签肽,如图3C所示,对于1 mg融合蛋白2 h即可完全除去标签肽。进一步,将酶切后的样品经镍亲和层析柱吸附6×His标签肽,收集穿透液再次超滤浓缩至6 mmol/L pH 7.0 Britton-Robinson缓冲液,调节超滤后ΔNspSEM融合蛋白的280 nm波长处吸光度使之与超滤前一致,SDS-PAGE表明(图3C泳道5),除去His标签肽的电泳纯ΔNspSEM重组蛋白纯度大于95%,满足后续光谱学测定要求。
2.3 纯化的His-ΔNspSEM质谱鉴定
图4 His-ΔNspSEM重组蛋白的质谱分析
Fig. 4 Identification of purified His-ΔNspSEM recombinant fusion protein by mass spectrometry
纯化的融合蛋白通过气相色谱-串联质谱鉴定,图4A表明,离子得分大于28的肽片段可信,图4B说明通过与金葡菌基因组数据库比对,鉴定的融合蛋白得分(大于75 000)与其他可能的蛋白得分相差10 000 倍以上,图4C显示,纯化的蛋白与来自金黄色葡萄球菌的M型肠毒素(NCBI编号:D0K667)氨基酸覆盖率在扣除了信号肽之后,高达96.3%,图4D是随机选择的一段肽段的指纹图谱。质谱鉴定表明,目标蛋白纯化成功。
2.4 纯化的ΔNspSEM重组蛋白圆二色谱与荧光发射谱测定
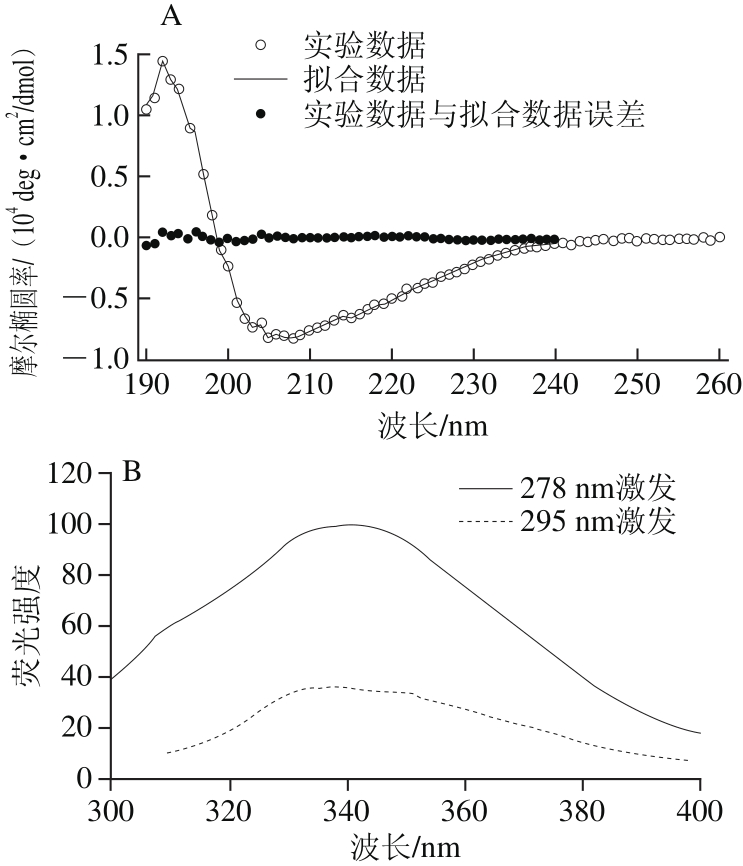
图5 ΔNspSEM重组蛋白远紫外圆二色谱(A)与室温荧光发射谱(B)
Fig. 5 Far-UV circular dichroism spectrum and room temperature fluorescence emission spectrum of ΔNspSEM fusion protein
图5 A所示为ΔNspSEM重组蛋白的远紫外区圆二色谱,表现为192 nm强正峰和208 nm强负峰信号,215 nm与222 nm波长处肩峰使负峰展宽。采用DichroWeb在线分析软件,计算出溶液中ΔNspSEM重组蛋白二级结构含量为:α-螺旋16%、β-折叠35%、β-转角21%和无规卷曲29%,实验数据与拟合数据之间的误差在可接受范围之内。图5B所示为ΔNspSEM重组蛋白的荧光发射谱,表现为278 nm激发和295 nm激发时,具有相同的341 nm色氨酸荧光发射峰。色氨酸的荧光发射峰位相对于亲水环境中的354 nm发射峰蓝移了13 nm,说明ΔNspSEM重组蛋白中色氨酸并未完全暴露于极性水环境中,重组蛋白具有折叠态的三级结构。
2.5 纯化的ΔNspSEM重组蛋白热稳定性分析
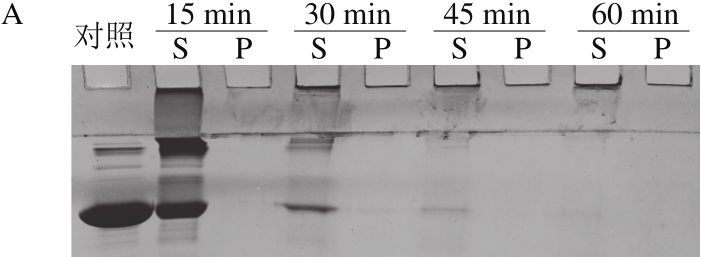
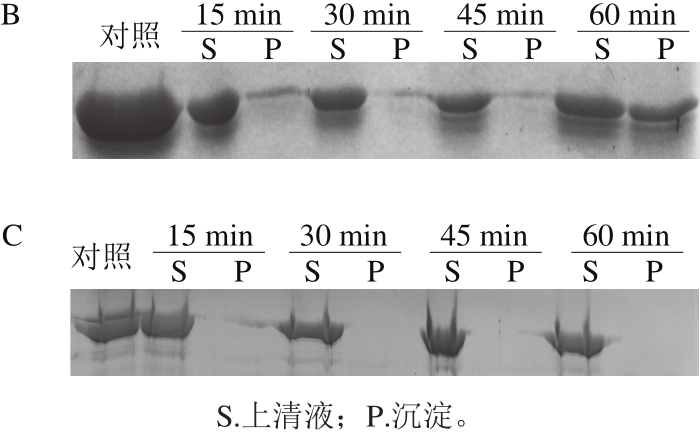
图6 100 ℃热处理对BSA(A)、SEM(B)和Pfu DNA Polymerase(C)稳定性的影响
Fig. 6 SDS-PAGE analysis of the effect of incubation at 100 ℃ on stability of BSA, SEM and Pfu DNA polymerase
将去除His标签肽的ΔNspSEM重组蛋白100 ℃热处理,间隔15 min采样,将离心所得上清液和沉淀分别进行SDS-PAGE分析。如图6所示,15~45 min范围内,主要在上清液中检测到ΔNspSEM重组蛋白,同时在沉淀中出现少量重组蛋白;经过1 h热处理后,沉淀中重组蛋白显著增加,但上清液和沉淀中重组蛋白总和基本不变。上述结果表明,经过100 ℃热处理1 h后,ΔNspSEM重组蛋白并未发生显著降解,而是产生了沉淀。对比溶解于相同缓冲液的BSA而言,经过15 min热处理,BSA形成大量可溶性高分子质量聚合体(有些聚合体分子质量高到沉积于加样孔,而不能进入4%浓缩胶内);30 min热处理后,可溶性BSA及其聚合体显著降解,同时在热处理过程中基本不产生沉淀。与之形成鲜明对比的是,商品化高保真DNA聚合酶(Pfu DNA Polymerase)在100 ℃加热1 h过程中,既不降解也不形成沉淀,具有很高热稳定性,这与该酶用于PCR循环过程中,需要94 ℃变性双链DNA所需高热稳定性要求是一致的。
本研究首先采用经典的BL21(DE3)原核表达系统[20-22],对His-ΔNspSEM蛋白编码的基因进行诱导表达。SDSPAGE表明目标蛋白表达量极低,因此,改用能有效抑制外源基因本底水平表达的BL21(DE3)pLysS系统[23-25],虽然实现了His-ΔNspSEM蛋白原核表达,但表达量较低。由此推测,BL21(DE3)原核表达系统对融合蛋白表达量低的原因是外源基因本底水平转录和表达降低了宿主系统的蛋白生产能力。进一步利用http∶//www.doe-mbi.ucla.edu/~sumchan/caltor.html在线分析,发现ΔNspSEM蛋白编码基因存在21个稀有密码子,分别为编码精氨酸的AGG(1 个)和AGA(4 个)、编码甘氨酸的GGA(9 个)、编码异亮氨酸的AUA(5 个)和编码亮氨酸的CUA(2 个)。稀有密码子的存在可能是造成表达量低的原因之一,为了提高融合蛋白表达量,将编码His-ΔNspSEM蛋白的基因转化到能够高效表达稀有密码子的Rosetta(DE3)菌株[26-27]中,由于该型菌株补充了上述5种稀有密码子对应的tRNA,目标蛋白的表达量显著提高。
通过圆二色谱分析揭示,ΔNspSEM重组蛋白溶液构象β-折叠为35%。王小红[28]研究表明,B型肠毒素具有高达40.4%的β-折叠,经过121 ℃处理30 min后,仍具有39.9%的β-折叠含量,表明高β-折叠对B型肠毒素热稳定性维持具有关键作用。本研究发现M型肠毒素与B型高度同源,因此可以推测ΔNspSEM重组蛋白溶液构象及维持也依赖于其高含量的β-折叠。
对ΔNspSEM重组蛋白的荧光发射谱进行分析,发现278 nm波长激发时未见显著的303 nm酪氨酸荧光发射峰,同时呈现特征性的341 nm色氨酸荧光发射主峰。该峰位与295 nm单独激发色氨酸残基时,产生的荧光发射峰接近,说明重组蛋白分子中酪氨酸吸收的能量通过荧光共振能量转移给色氨酸[29],形成了341 nm的色氨酸荧光发射峰。因此,根据蛋白质分子内能量转移有效性和色氨酸荧光峰位相对于极性水环境(354 nm发射峰)的显著蓝移[30],可以推测纯化的重组蛋白处于构象紧密的天然折叠状态。
分别以BSA和Pfu DNA Polymerase为100 ℃热处理负对照和正对照,ΔNspSEM重组蛋白表现出较高的热稳定性,这与天然SEM具有类似的性质。
综上,本研究成功构建了高产融合蛋白His-ΔNspSEM的可溶性原核表达体系,经该表达体系纯化并切除标签肽之后的重组蛋白溶液构象紧密,其溶液构象的维持依赖于二级结构中的高含量的β-折叠,这为进一步研究SEM的结构与功能关系提供了理论支持。
参考文献:
[1] DEURENBERG R H, STOBBERINGH E E. The evolution of Staphylococcus aureus[J]. Infection Genetics and Evolution, 2008,8(6): 747-763. DOI:10.1016/j.meegid.2008.07.007.
[2] DAVID M Z, DAUM R S. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic[J]. Clinical Microbiology Reviews, 2010, 23(3):616-687. DOI:10.1128/CMR.00081-09.
[3] KHORAMROOZ S S, MANSOURI F, MARASHIFARD M, et al.Detection of biofilm related genes, classical enterotoxin genes and agr typing among Staphylococcus aureus isolated from bovine with subclinical mastitis in southwest of Iran[J]. Microbial Pathogenesis,2016, 97: 45-51. DOI:10.1016/j.micpath.2016.05.022.
[4] KING J M, KULHANKOVA K, STACH C S, et al. Phenotypes and virulence among Staphylococcus aureus USA100, USA200, USA300,USA400, and USA600 clonal lineages[J]. mSphere, 2016, 1(3):e00071-16. DOI:10.1128/mSphere.00071-16.
[5] MCCORMICK J K, YARWOOD J M, SCHLIEVERT P M. Toxic shock syndrome and bacterial superantigens: an update[J]. Annual Review of Microbiology, 2001, 55: 77-104. DOI:10.1146/annurev.micro.55.1.77.
[6] DINGES M M, ORWIN P M, SCHLIEVERT P M. Exotoxins of Staphylococcus aureus[J]. Clinical Microbiology Reviews, 2000,13(1): 16-34. DOI:10.1128/CMR.13.1.16-34.2000.
[7] BALABAN N, RASOOLY A. Staphylococcal enterotoxins[J].International Journal of Food Microbiology, 2000, 61(1): 1-10.DOI:10.1016/S0168-1605(00)00377-9.
[8] 王琼, 唐俊妮. 金黄色葡萄球菌肠毒素及移动基因元件研究进展[J]. 食品科学, 2016, 37(3): 241-246. DOI:10.7506/spkx1002-6630-20160342.
[9] ORWIN P M, FITZGERALD J R, LEUNG D Y, et al. Characterization of Staphylococcus aureus enterotoxin L[J]. Infection and Immunity,2003, 71(5): 2916-2919. DOI:10.1128/IAI.71.5.2916-2919.2003.
[10] ORWIN P M, LEUNG D Y, DONAHUE H L, et al. Biochemical and biological properties of staphylococcal enterotoxin K[J]. Infection and Immunity, 2001, 69(1): 360-366. DOI:10.1128/IAI.69.1.360-366.2001.
[11] ORWIN P M, LEUNG D Y, TRIPP T J, et al. Characterization of a novel staphylococcal enterotoxin-like superantigen, a member of the group V subfamily of pyrogenic toxins[J]. Biochemistry, 2002, 41(47):14033-14040. DOI:10.1021/bi025977q.
[12] GÜNTHER S, VARMA A K, MOZA B, et al. A novel loop domain in superantigens extends their T cell receptor recognition site[J].International Journal of Biological Macromolecules, 2007, 371(1):210-212. DOI:10.1016/j.jmb.2007.05.038.
[13] LINA G, BOHACH G A, NAIR S P, et al. Standard nomenclature for the superantigens expressed by Staphylococcus[J]. Journal of Infectious Diseases, 2004, 189(12): 2334 -2336. DOI:10.1086/420852.
[14] DINGES M M, ORWIN P M, SCHLIEVERT P M. Exotoxins of Staphylococcus aureus[J]. Clinical Microbiology Reviews, 2000,13(1): 16-34. DOI:10.1128/CMR.13.1.16-34.2000.
[15] EVENSON M L, HINDS M W, BERNSTEIN R S. Estimation of human dose of staphylococcal enterotoxin A from a large outbreak of staphylococcal food poisoning involving chocolate milk[J].International Journal of Food Microbiology, 1988, 7(4): 311-316.DOI:10.1016/0168-1605(88)90057-8.
[16] OMOE K, HU D L, ONO H K, et al. Emetic potentials of newly identified staphylococcal enterotoxin-like toxins[J]. Infection and Immunity, 2013, 81(10): 3627-3631. DOI:10.1128/IAI.00550-13.
[17] JARRAUD S, PEYRAT M A, LIM A. Egc, a highly prevalent operon of enterotoxin gene, forms a putative nursery of superantigens in Staphylococcus aureus[J]. Journal of Immunology, 2001, 166(1): 669-677. DOI:10.4049/ jimmunol.166.1.669.
[18] ZHAO Y, ZHU A N, TANG J, et al. Identification and measurement of staphylococcal enterotoxin-like protein Ⅰ (SEll) secretion from Staphylococcus aureus clinical isolate[J]. Journal of Applied Microbiology, 2016, 121(2): 539-546. DOI:10.1111/jam.13181.
[19] ABDUL-GADER A, MILES A J, WALLACE B A. A reference dataset for the analyses of membrane protein secondary structures and transmembrane residues using circular dichroism spectroscopy[J].Bioinformatics, 2011, 27(12): 1630-1636. DOI:10.1093/bioinformatics/btr234.
[20] SØRENSEN H P, MORTENSEN K K. Advanced genetic strategies for recombinant protein expression in Escherichia coli[J].Journal of Biotechnology, 2011, 115(2): 113-128. DOI:10.1016/j.jbiotec.2004.08.004.
[21] LIU J, XIE S S, LUO Y, et al. Soluble expression of spinach psbC gene in Escherichia coli and in vitro reconstitution of CP43 coupled with chlorophyll a only[J]. Plant Physiology and Biochemistry, 2014,79: 19-24. DOI:10.1016/j.plaphy.2014.02.026.
[22] TAVAKOLI A, HAMZAH A, RABU A. Expression, purification and kinetic characterization of recombinant benzoate dioxygenase from Rhodococcus ruber UKMP-5M[J]. Molecular Biology Research Communications, 2016, 5(3): 133-142.
[23] FARHANGNIA L, GHAZNAVI-RAD E, MOLLAEE N, et al.Cloning, expression and purification of recombinant lysostaphin from Staphylococcus simulans[J]. Jundishapur Journal of Microbiology,2014, 7(5): e10009. DOI:10.5812/jjm.10009.
[24] JIA B, JEON C O. High-throughput recombinant protein expression in Escherichia coli: current status and future perspectives[J]. Open Biology, 2016, 6(8). DOI:10.1098/rsob.160196.
[25] PAN S H, MALCOLM B A. Reduced background expression and improved plasmid stability with pET vectors in BL21(DE3)[J].Biotechniques, 2000, 29(6): 1234-1238.
[26] WANG N, REN K, JIA R. Expression of a fungal manganese peroxidase in Escherichia coli: a comparison between the soluble and refolded enzymes[J]. BMC Biotechnology, 2016, 16(1): 87-102.DOI:10.1186/s12896-016-0317-2.
[27] OHMORI T, MORITA H, TANAKA M. Expression in Escherichia coli of biphenyl 2,3-dioxygenase genes from a Gram-positive polychlorinated biphenyl degrader, Rhodococcus jostii RHA1[J].Bioscience Biotechnology and Biochemistry, 2011, 75(1): 26-33.DOI:10.1271/bbb.100452.
[28] 王小红. 金黄色葡萄球菌B型肠毒素的溶液构象及菌体生长环境效应[D]. 武汉: 华中农业大学, 2005.
[29] BOTEVA R, ZLATEVA T, DOROVSKA-TARAN V. Dissociation equilibrium of human recombinant interferon gamma[J]. Biochemistry,1996, 35(47): 14825-14830. DOI:10.1021/bi9527597.
[30] ROYER C A. Probing protein folding and conformational transitions with fluorescence[J]. Chemical Reviews, 2006, 106(5): 1769-1784.DOI:10.1021/cr0404390.
Prokaryotic Expression, Purification, Identification and Solution Conformation of Staphylococcal Enterotoxin M
LIU Ji, YANG Fan, TIAN Wanfan, LONG Hu, SUN Siyu, ZHOU Yushan, ZHAO Yanying, TANG Junni*
(College of Life Science and Technology, Southwest Minzu University, Chengdu 610041, China)
Abstract:Staphylococcal enterotoxin M (SEM) is a secretory superantigen encoded by the νSa genomic islands of Staphylococcus aureus. In this study, the sem gene from S. aureus without N-terminal signal peptide was subcloned into the prokaryotic expression vector pET-28a (+) to construct the recombinant plasmid pET-28a (+)-ΔNspsem and the recombinant expression plasmid was then transformed into E. coli Rosetta (DE3) competent cells. The positive clones, induced by IPTG, effectively expressed His-tag containing soluble ΔNspSEM fusion protein in E. coli Rosetta (DE3). The expression conditions including expression vector, time, IPTG concentration and temperature were optimized. Purified His-ΔNspSEM fusion protein was obtained by Ni2+-Sepharose affinity chromatography. Mass spectrometric analysis indicated that the amino acid sequence of the fusion protein was 96.3% similar to that of SEM. Circular dichroism revealed ΔNspSEM, whose 6 × His sequence was cleaved by thrombin, was rich in β-sheet (35%) and β-turn (21%) but low in α-helix (16%). The fluorescence emission spectrum of ΔNspSEM exhibited identical tryptophan emission peak (341 nm) with excitation at 278 and 295 nm.The time-dependent thermal stability of ΔNspSEM obtained at 100 ℃ by SDS-PAGE indicated that the recombinant protein had relatively high thermal stability. To conclude, our results showed that the ΔNspSEM recombinant protein was successfully expressed and that the purified protein exhibited a compact conformation similar to the natural one in solution,which can provide a basis for insight into the structure and function of SEM protein.
Key words:staphylococcal enterotoxin M; prokaryotic expression; purification; mass spectrometry; circular dichroism;fluorescence emission spectroscopy; thermal stability
DOI:10.7506/spkx1002-6630-201724007
中图分类号:TS201.1
文献标志码:A
文章编号:1002-6630(2017)24-0040-07
收稿日期:2016-11-12
基金项目:国家自然科学基金面上项目(31371781);四川省应用基础项目(2014JY0253);
西南民族大学大学生创新创业训练计划项目(201610656053);四川省教育厅项目(15ZB0482)作者简介:刘骥(1978—),男,讲师,博士,研究方向为食品与生物技术。E-mail:liuji965@aliyun.com
*通信作者:唐俊妮(1971—),女,教授,博士,研究方向为食品安全与食品微生物。E-mail:junneytang@aliyun.com
引文格式:
刘骥, 杨帆, 田万帆, 等. 金黄色葡萄球菌M型肠毒素原核表达、纯化、鉴定及溶液构象分析[J]. 食品科学, 2017,38(24): 40-46.
DOI:10.7506/spkx1002-6630-201724007. http∶//www.spkx.net.cn
LIU Ji, YANG Fan, TIAN Wanfan, et al. Prokaryotic expression, purification, identification and solution conformation of staphylococcal enterotoxin M[J]. Food Science, 2017, 38(24)∶ 40-46. (in Chinese with English abstract) DOI∶10.7506/spkx1002-6630-201724007. http∶//www.spkx.net.cn