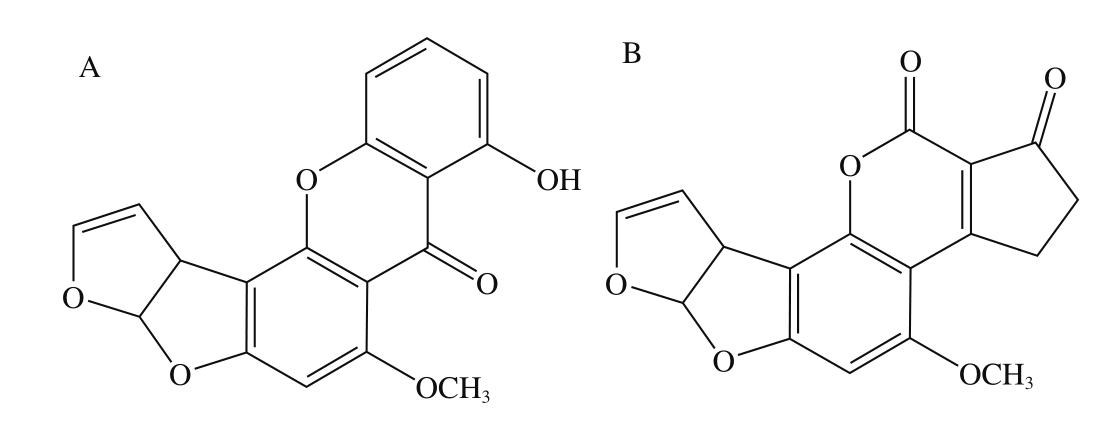
图1 STC(A)和黄曲霉毒素B1(B)化学结构式
Fig. 1 Chemical structures of sterigmatocystin (A) and aflatoxin B1(B)
周 健1,2,许娇娇1,*,金米聪2,蔡增轩1,黄百芬1,任一平3
(1.浙江省疾病预防控制中心理化毒理所,浙江 杭州 310051;2.宁波市疾病预防控制中心,浙江省微量有毒化学物健康风险评估技术研究重点实验室,浙江 宁波 315010;3.国家食品安全风险评估中心应用技术合作中心,浙江清华长三角研究院,浙江 嘉兴 314006)
摘 要:使用乙腈溶液对鸡蛋中杂色曲霉毒素进行提取,后加入无水Na2SO4、NaCl和无水CH3COONa进行盐析,取出上层乙腈后加入C18吸附剂和无水Na2SO4进行净化浓缩后上机检测。采用Plackett-Burman试验、单因素试验和响应面法优化,最大限度地提高杂色曲霉毒素的提取率。使用基质匹配曲线外标法定量,所有阳性样品均使用免疫亲和柱法复测。结果表明,杂色曲霉毒素在0.125~1 000 ng/mL质量浓度范围内线性良好,相关系数达0.999 6,检出限为0.1 μg/kg,定量限为0.5 μg/kg。在空白鸡蛋基质中进行三水平加标实验,测得提取率在86.8%~90.4%范围内,日间重复性在1.5%~6.2%范围内。最终,将建立的方法用于45 份样品检测,其中10 份鸡蛋样品检测结果呈阳性,含量为0.5~3 608 μg/kg。
关键词:鸡蛋;杂色曲霉毒素;QuEChERS法;响应面法;超高效液相色谱-质谱联用
鸡蛋是人类最重要的动物源性食物之一,富含蛋白质、胆固醇及矿物质等营养成分,有利于促进人体生长、预防疾病、改善健康状况等,并且常常被加工成其他各类食品。近几年随着社会经济的发展,生活质量的提高,食品中有毒有害物质尤其是真菌毒素污染已成为政府重视、社会关注、世界瞩目的热点问题[1-3]。为保障食品安全,降低人群摄入真菌毒素的风险,各国对食品(主要针对谷物及其衍生产品)中部分真菌毒素规定了相应的残留限量标准,但鲜有研究调查鸡蛋中真菌毒素的污染水平[4-6],而对鸡蛋中杂色曲霉毒素(sterigmatocystin,STC)的检测报道更是少之又少。
STC主要由杂色曲霉(Aspergillus versicofor)、构巢曲霉(A. nidulans)、黄曲霉(A. flavus)和皱曲霉(A.rugulosus)等产毒真菌代谢生成,广泛分布于自然界中,可污染大米、小麦、玉米、豆类、花生饼及饲料[7-8]。被污染的粮食及饲草被动物摄入后,会通过食物链进入人类体内,从而对人体健康造成损害[9-10]。STC是第一个被发现含有双氢呋喃苯并呋喃体系的天然产物,其结构与黄曲霉毒素B1十分相似,两者结构式如图1所示。通过14C同位素标记法已证实,STC可以转变为黄曲霉素B1,其毒性在目前已知的真菌毒素中仅次于黄曲霉素B1,致癌能力约为黄曲霉素B1的1/10,此外,STC还具有肾、肝毒性,会导致动物肝硬变、中毒性肝炎、肝细胞和肾小管上皮细胞坏死等[11]。
图1 STC(A)和黄曲霉毒素B1(B)化学结构式
Fig. 1 Chemical structures of sterigmatocystin (A) and aflatoxin B1(B)
对食品中真菌毒素测定的预处理过程主要包括提取和净化2 个步骤,而食品基质通常成分复杂,因此净化步骤成为分析实验关键步骤[12]。目前真菌毒素的主要净化方法是在固相萃取法基础上衍生的免疫亲和柱法[12-14]和多功能柱法[15-17],前者虽然净化效果较好,但存在成本过高、耗时长、无法同时测定多组分等缺陷;后者处理样品速度快,适用范围广,但净化效果有限,部分待测毒素的绝对提取率较低。除固相萃取法外,QuEChERS法因其快速、简单、廉价、高效、稳定、安全、样品通量高等特点,近年来被广泛应用于真菌毒素检测[18-20],但QuEChERS法净化过程往往涉及到较多可变的实验条件,如果采用传统的单因素试验法则耗时耗力,且无法对条件间相互作用进行评估[21-22]。
本实验采用QuEChERS法对鸡蛋中的STC进行提取与净化,结合Plackett-Burman设计对前处理条件中显著性因素进行筛选,单因素试验优化确定关键因素最佳范围,随后使用响应面中心组合试验对待测因素进行试验设计与优化,建立STC提取率的拟合模型并找出最佳实验条件。最后在最佳条件下进行方法学验证,并应用于实际样品检测。
1.1 材料与试剂
新鲜鸡蛋为农贸市场及超市中随机采购的样品,同一批次样品视为一份,样品用均质机均质后于-24 ℃冷藏备用。
STC标准品(纯度≥98%) Romer国际贸易(北京)有限公司;STC免疫亲和柱 德国R-Biopharm公司;甲醇、乙腈(色谱纯) 德国Merck公司;丙基乙二胺(primary secondary amine,PSA)吸附剂、C18吸附剂 上海安谱实验科技股份有限公司;DisQuE净化盐包 美国Waters公司;无水MgSO4、无水Na2SO4、无水CH3COONa、NaCl(分析纯)(除无水CH3COONa外其他盐类使用前均在300 ℃恒温4 h) 上海凌峰化学试剂有限公司。
STC标准储备溶液:取STC标准品1.00 mg,用乙腈溶解并定容至10.0 mL,最终质量浓度为100 μg/mL;标准工作溶液用乙腈将储备溶液稀释至所需质量浓度。
1.2 仪器与设备
Waters AcquityTM超高效液相色谱配Xevo TQ-S串联质谱 美国Waters公司;Milli-Q超纯水仪 美国Millipore公司。
1.3 方法
1.3.1 样品前处理
QuEChERS方法参考Xu Jiaojiao[20]和Zhou Jian[23]等并作适当修改:称取2.50 g鸡蛋于50 mL离心管中,再加入5.0 mL水,室温振荡2 min后加入10.0 mL乙腈,充分混合均匀后超声萃取30 min。萃取后加入4.0 g无水Na2SO4,1.43 g NaCl和0.95 g无水CH3COONa进行盐析,振荡1 min,8 500 r/min离心3 min,取3.0 mL上层乙腈加入300 mg C18吸附剂和1.5 g无水Na2SO4混合净化,吸取1.0 mL提取液氮吹浓缩,最后使用1.0 mL 20%乙腈溶液复溶后过0.22 μm滤膜上机分析。
阳性样品处理按照GB 5009.25—2016《食品中杂色曲霉素的测定》中第二法操作,并根据样品中毒素含量对提取液稀释比例作适当调整以免超过净化柱载荷量。
1.3.2 QuEChERS盐析
在QuEChERS提取过程中,盐析分层是直接影响待测物提取率的关键步骤。经典QuEChERS萃取法通常使用无水MgSO4和NaCl来进行盐析[24-25],但在实际操作过程中发现无水MgSO4在吸水时放热剧烈并迅速结块,可能会导致目标物被包裹,同时,瞬间升温也可能促使某些热不稳定物质分解。本实验考察MgSO4、Na2SO4、CH3COONa、NaCl等在QuEChERS提取中对STC提取率的影响,并设计以下6 种组合:4.0 g MgSO4+1.0 g CH3COONa、4.0 g MgSO4+1.0 g NaCl、4.0 g MgSO4+1.0 g NaCl+1.0 g Na3C6H5O7+0.5 g Na2C6H6O7、4.0 g CH3COONa+1.0 g NaCl、4.0 g Na2SO4+1.0 g NaCl、4.0 g Na2SO4+1.0 g CH3COONa。以50 μg/kg添加量对空白鸡蛋基质进行加标,取3.0 mL上清液用500 mg C18吸附剂进行净化后浓缩过膜进样,每种盐类组合进行3 次平行实验。
1.3.3 Plackett-Burman试验与单因素试验
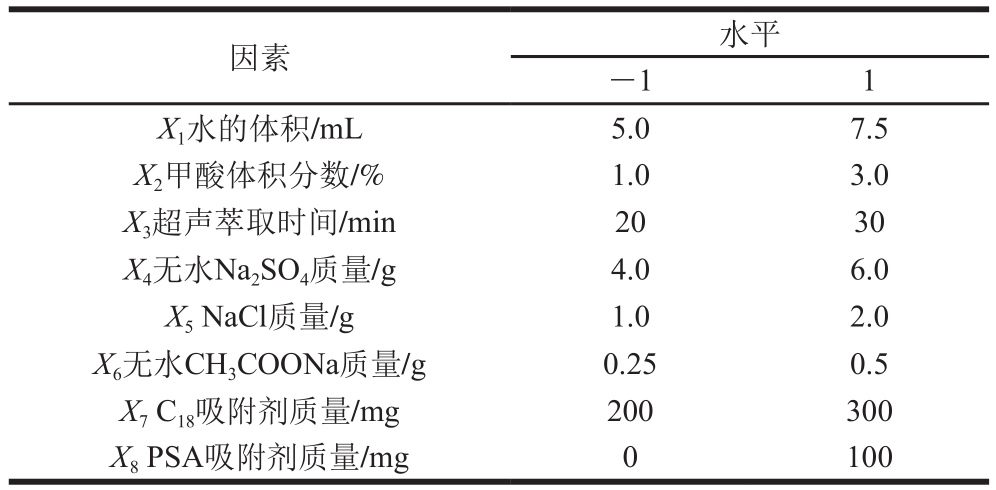
在确定最佳QuEChERS盐类组合后,采用Plackett-Burman试验(n=20),对QuEChERS萃取中水体积、乙腈中甲酸体积分数、超声萃取时间、无水Na2SO4质量、NaCl质量、无水CH3COONa质量、吸附剂(C18和PSA)质量8 个因素进行考察,试验设计如表1所示,其中-1代表低水平,1代表高水平,响应值为3 次平行试验STC提取率的平均值。
表1 Plackett-Burman试验设计因素与水平
Table 1 Coded levels and corresponding actual levels of independent variables used for Plackett-Burman design
根据Plackett-Burman试验结果,对筛选出的关键因素(PSA、NaCl、无水CH3COONa质量以及甲酸体积分数)进行单因素控制变量试验,以缩小后续响应面试验优化范围。
1.3.4 中心组合试验设计
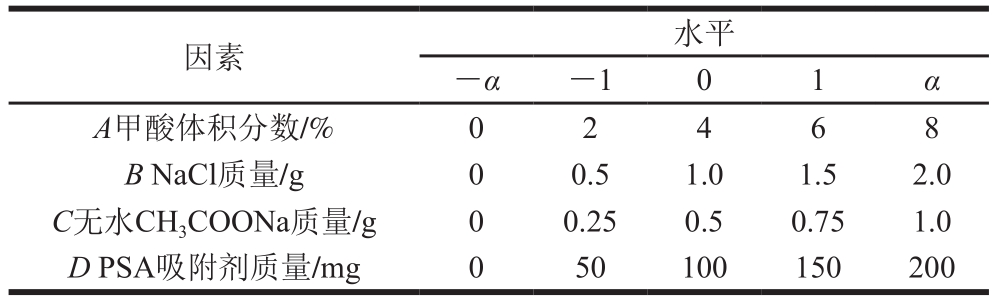
在确定每个待优化因素大致的最佳范围后,对其进行中心组合响应面设计,每个因素进行5 个不同水平的试验来减少操作中偶然误差的影响,试验设计因素与水平如表2所示。
表2 中心组合试验设计因素与水平
Table 2 Coded levels and corresponding actual levels of independent variables used for central composite design
利用软件对试验结果进行回归拟合后,可得到试验因素的二次多项回归模型,见公式(1):
式中:Y为预测响应值,Xi和Xj代表独立变量,δ0为常数项,δi为线性系数,δii为二次项系数,δij为交互项系数,ε为随机误差补偿项。
1.3.5 高效液相色谱-质谱检测条件
液相色谱条件:采用Waters CORTECSTM色谱柱(150 mm ×3.0 mm,1.6 μm),柱温40 ℃,流速0.4 mL/min,进样体积10 μL,流动相A:体积分数0.1%甲酸溶液,流动相B:0.1%甲酸-乙腈溶液。梯度洗脱条件:0~0.8 min,10% B;0.8~2.0 min,10%~20% B;2.0~7.0 min,20% B;7.5~10.5 min,20%~40% B;10.5~11.0 min,40%~60% B;11.0~14.0 min,60% B;14.0~14.2 min,60%~95% B;14.2~16.2 min,95% B;16.2~16.5 min,95%~10% B;16.5~20.0 min,10% B。
质谱条件:电喷雾离子源(electrospray ionization,ESI),正ESI多反应监测模式,毛细管电压3.0 kV,离子源温度500 ℃,脱溶剂气(高纯氮气,纯度>99%)流速750 L/h,碰撞气(高纯氩气,纯度>99%)流速0.15 mL/min。STC质谱参数:锥孔电压40 V,母离子m/z 325.09,定性子离子m/z 310.06,定量子离子m/z 281.08,碰撞能量分别为23 eV和32 eV。
1.3.6 基质效应评估与方法学验证
基质共萃取物会在离子源(尤其在ESI源)中与待测物质竞争发生离子化反应,这也通常导致待测物离子化效率降低,产生基质抑制效应。本实验中使用提取后加入法来评估鸡蛋样品基质效应,具体操作步骤:在纯溶剂将STC标准工作溶液稀释成7个不同质量浓度的溶液(0.125、0.25、0.5、2.5、10、50、250、1 000 ng/mL);在空白鸡蛋提取液浓缩复溶的同时加入相同质量浓度水平的标准溶液,将所得曲线的斜率进行对比即为绝对基质效应,一般绝对基质效应在80%~120%之间认为是可接受范围[26]。此外,不同批次的样品、采样时间以及喂养饲料成分的差异可能也会导致样品间内源性成分的不同,此时应评估基质的相对基质效应:取5 份不同的空白样品,进行绝对基质效应测试,所得基质效应值的相对标准偏差(relative standard deviation,RSD)小于15%时可认为样品间不存在较大差异[27]。
方法准确性主要通过三浓度水平加标实验来评估,每个加标水平实验平行6 次,精密度则通过日间、日内(连续重复3 d)重复性RSD来表示。取空白鸡蛋样品以1.0 μg/kg水平加标,平行重复6 次实验,进样后分别以3 倍和10 倍信噪比为方法检出限和定量限。
1.4 数据分析
仪器数据分析软件为Waters Mass Lynx 4.1和Target Lynx 4.1;实验设计及统计分析软件为Design-Expert 8.0.6.0,Minitab 17.1.0和IBM SPSS Statistics 19.0。试样中STC含量按公式(2)计算:
式中:X为试样中STC含量/(μg/kg);ρ为由标准曲线计算所得的试样溶液中STC质量浓度/(ng/mL);V为最终定容体积/mL;m为样品质量/g;f为稀释倍数。
2.1 QuEChERS法盐析优化结果
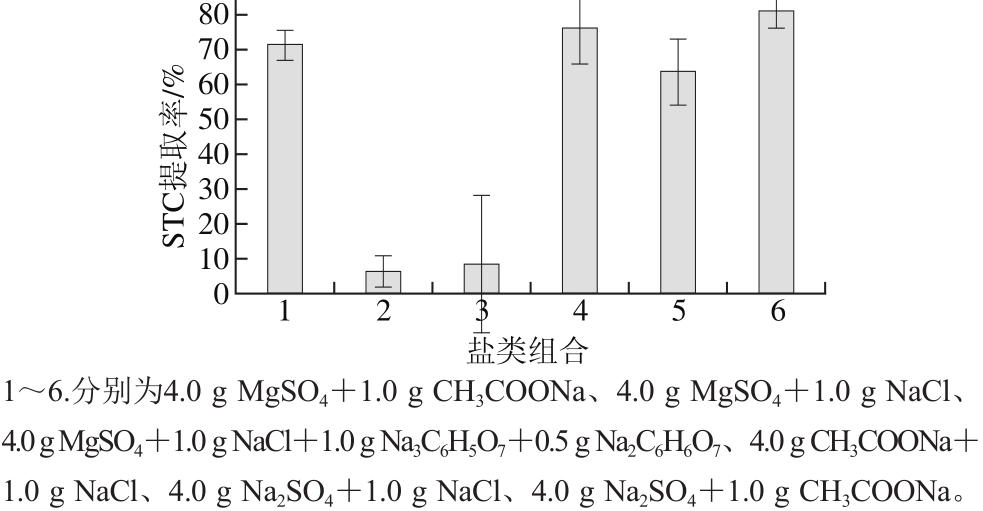
图2 盐类组合对STC提取率的影响
Fig. 2 Effects of salt combinations on the extraction efficiency of sterigmatocystin
由图2可知,使用无水Na2SO4和无水CH3COONa作为脱水剂组合均有较高的提取率,考虑到盐类吸水性能,最终选用无水Na2SO4作为脱水剂。根据组合4和6结果可知,盐析剂NaCl和CH3COONa两者效果较好,但其所用量需要进一步优化。
2.2 Plackett-Burman试验结果及分析
表3 Plackett-Burman试验设计结果
Table 3 Plackett-Burman design with response variable
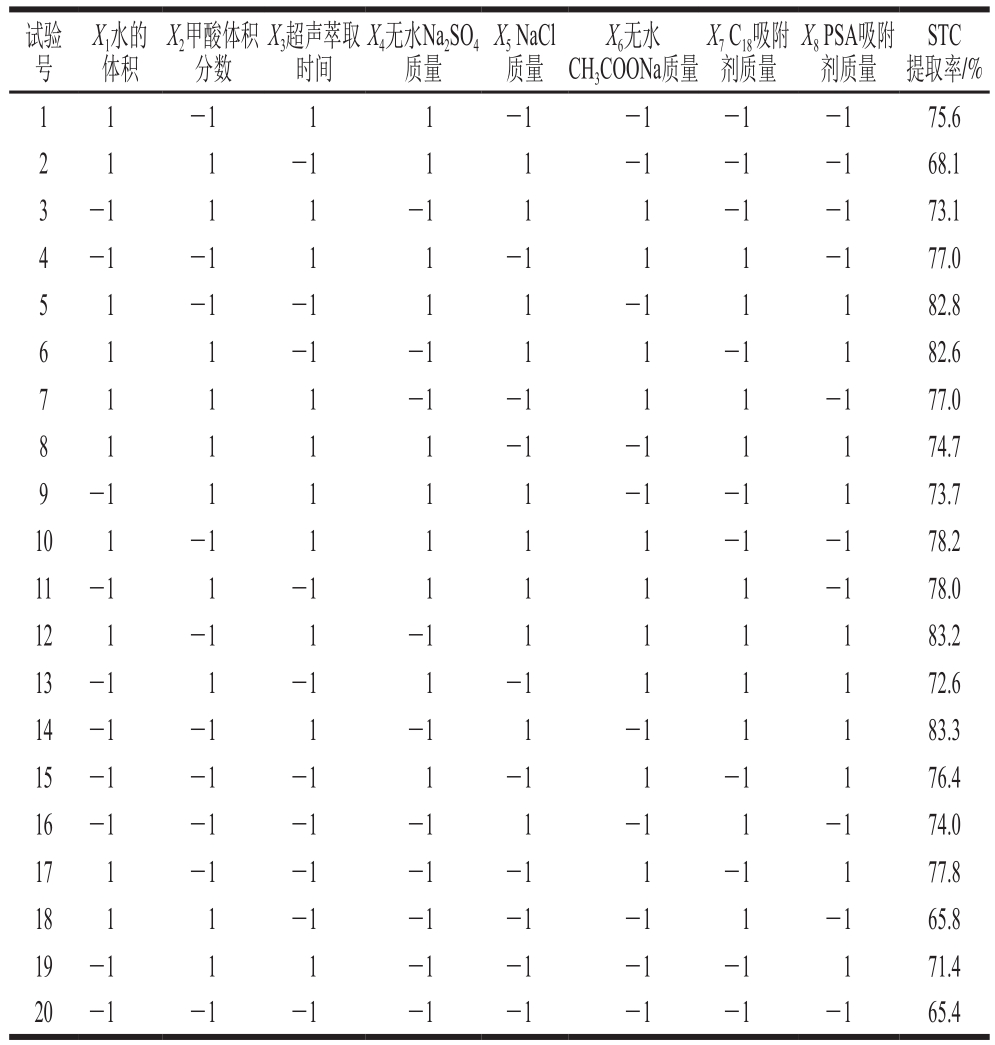
采用Plackett-Burman设计对实验中大部分条件进行初步筛选,基于非完全平衡块原理,能在实验次数较少的前提下选出对实验结果有显著影响的关键因素[28]。每个因素各取高低两水平,其中高水平通常设置为低水平的1.5 倍。通过对表3所得数据进行分析后,将结果用帕累托图表示,如图3所示,其中4 个因素的标准化效应值超过了基准参照线(图中虚线),表明这些因素对提取率影响显著,在后续试验中作为主要因素进一步优化。
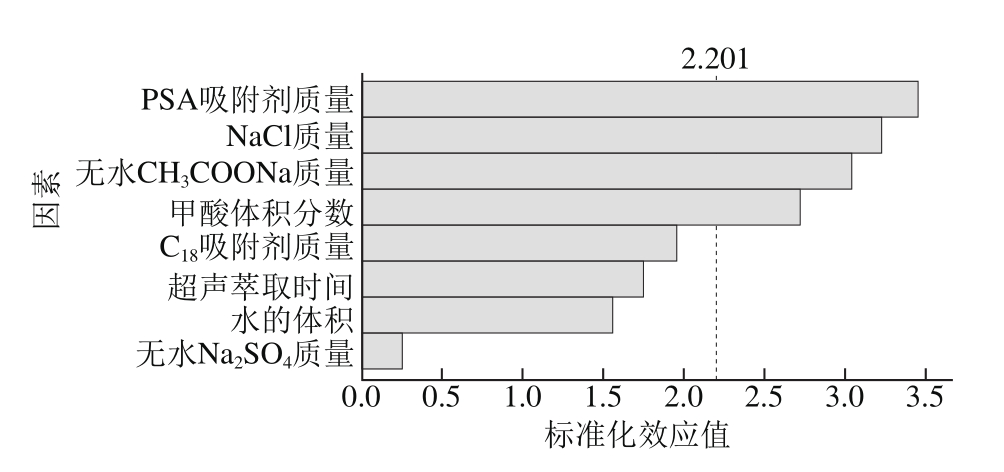
图3 试验因素标准化效应帕累托图
Fig. 3 Pareto chart of the standardized effects
2.3 单因素试验优化结果
单因素试验采用控制变量法,对各因素进行五水平考察,确定其最佳值所处范围供响应面法参考。根据Plackett-Burman试验结果,假设4 个显著性因素的中心水平为:PSA吸附剂质量100 mg、NaCl质量1.0 g、无水CH3COONa质量1.0 g、甲酸体积分数4%。因素的5 个考察水平分别为:PSA吸附剂质量0、50、100、150、200 mg;NaCl质量0、0.5、1.0、1.5、2.0 g;无水CH3COONa质量0、0.5、1.0、1.5、2.0 g;甲酸体积分数0%、2%、4%、6%、8%,当考察因素变化时其余因素均固定为中心水平,结果如图4所示。最终选择的条件为PSA吸附剂质量0 mg、NaCl质量1.0 g、无水CH3COONa质量0.5 g、甲酸体积分数0%。
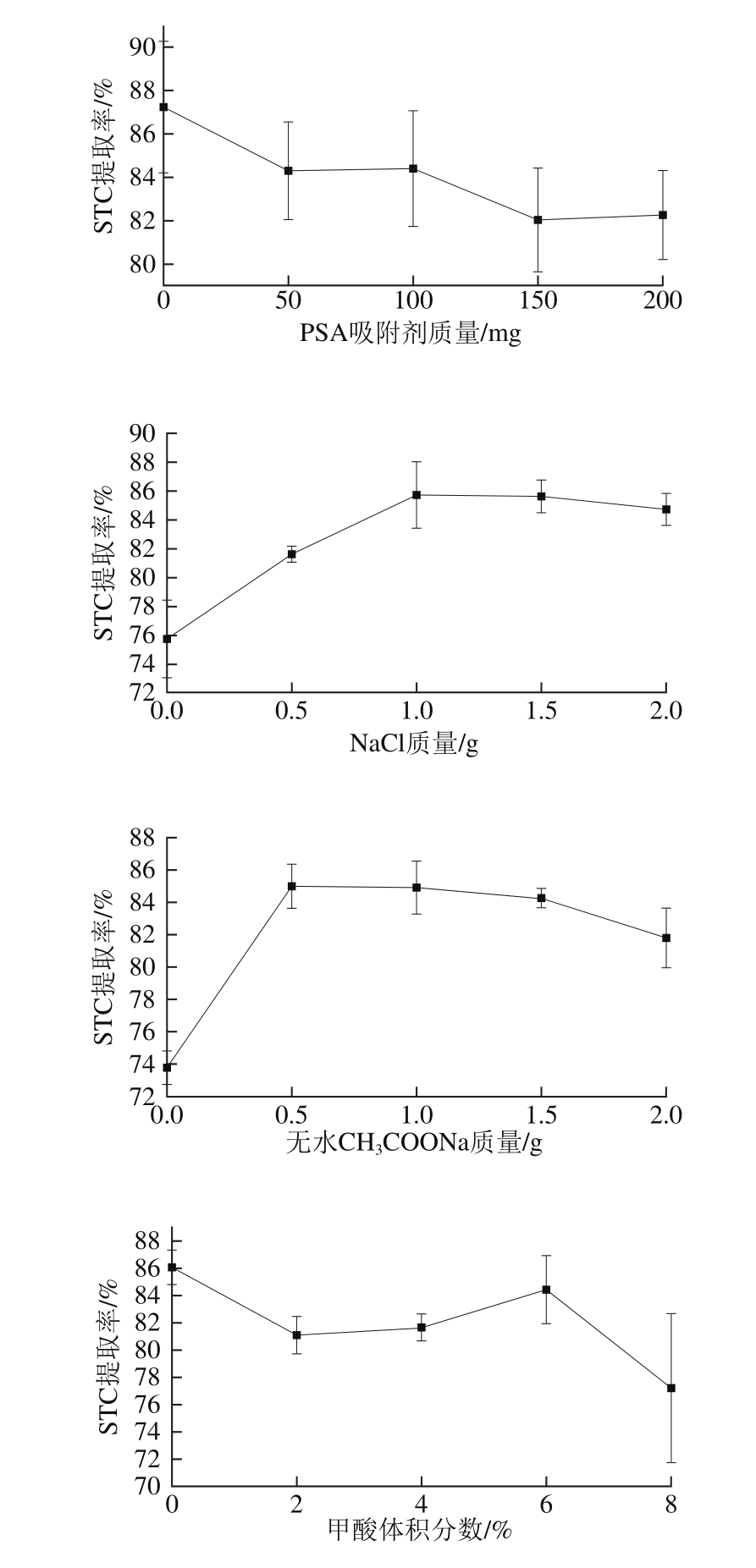
图4 各单因素对提取率的影响
Fig. 4 Individual effect of variables on the extraction efficiency
2.4 中心组合设计与响应面试验结果
根据Plackett-Burman试验和单因素优化试验确定的试验因素和水平,采用中心组合设计试验对加标鸡蛋进行四因素五水平的响应面分析(表4),以减少操作过程中偶然误差的影响。
表4 中心组合试验设计结果
Table 4 Central composite design with response variable
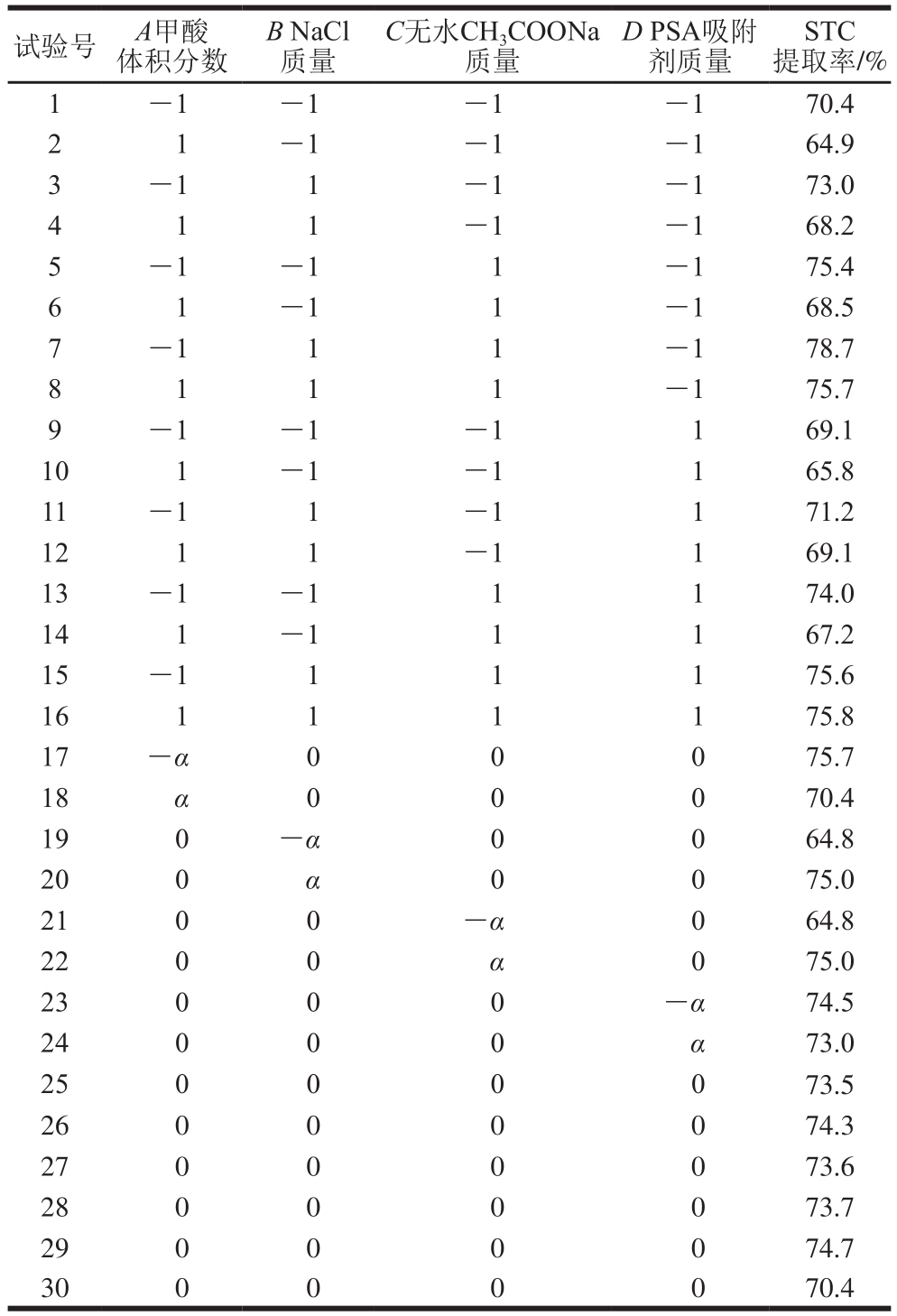
对表4中所得数据进行二次多元回归拟合,整理得到对试验因素一次项、交互项和二次项进行评估的回归方程如下:
STC提取率=73.77-1.78A+2.19B+2.48C-0.42D+0.80AB-0.047AC+0.52AD+0.58BC-0.044BD-0.28CD-0.22A2-1.01B2-1.00C2-0.038D2
对上述结果进行方差分析,表5显示该拟合方程模型项为极显著,失拟项为不显著,在模型系数项中,所有一次项除D项为显著水平外其余均为极显著水平,交互项中AB、AD和BC为显著,二次项B2、C2为极显著水平。多项式模型方程的拟合程度和模型质量由确定系数(R2)表示,模型拟合良好一般要求R2至少大于0.8[29],本试验中R2为0.969 9,调整确定系数(R2Adj)为0.941 9,表明生成的方程与试验数据拥有96.99%的符合度,能够解释94.19%的变化效应,对响应值的预测能力优秀,可信度较高[30]。
根据回归方程利用软件作不同因素交互项的响应面图(图5),可直观地反映出各因素交互作用对STC提取率的影响。在响应面图中2 个考察因素的变化范围从-α水平到α水平,而其他因素则维持在中心水平。
表5 二次方差模型的方差分析
Table 5 Analysis of variance of the quadratic equation model

注:P<0.01,差异极显著;P<0.05,差异显著。
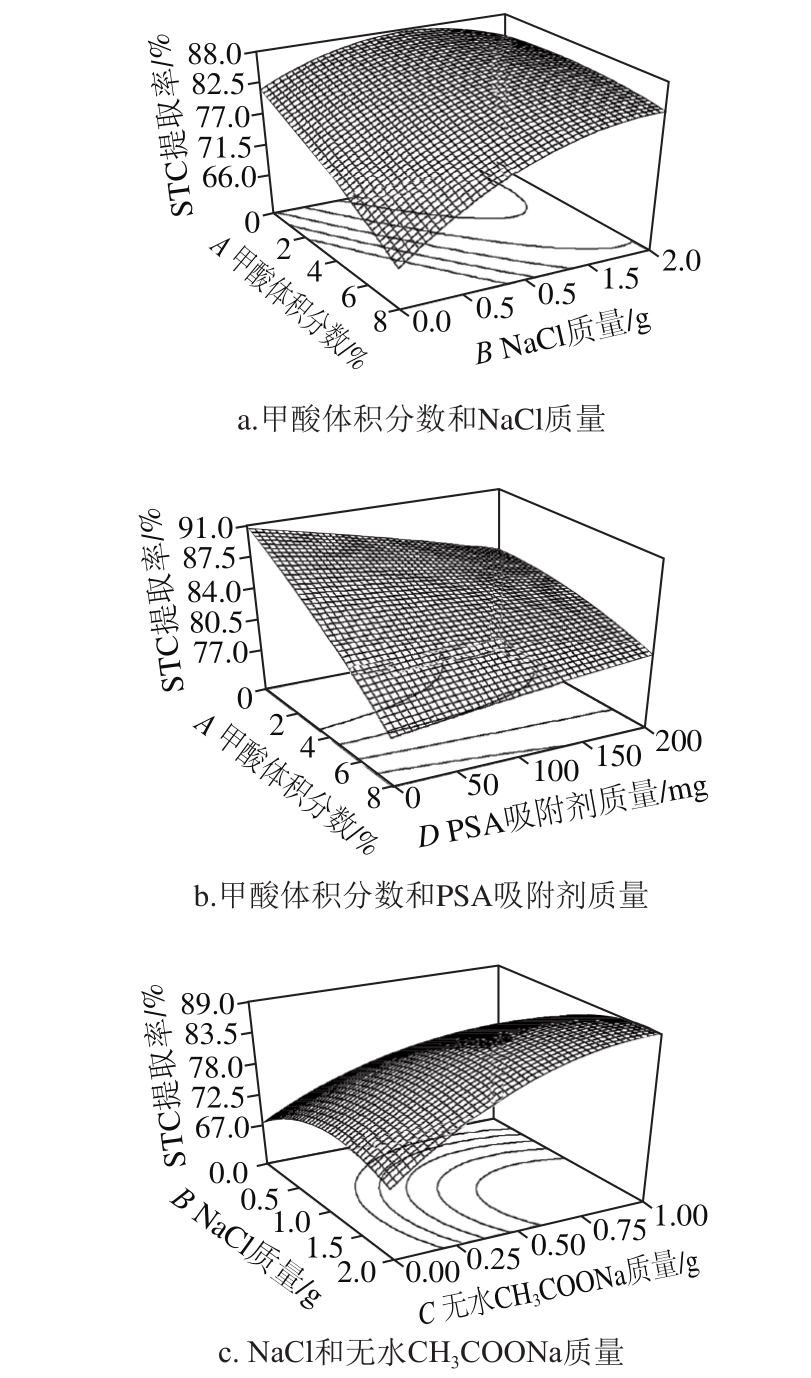
图5 各因素交互作用对鸡蛋中STC提取率影响的响应面图
Fig. 5 Response surface plots showing the interactive effect of variables on the extraction efficiency of sterigmatocystin
由图5a可知,当甲酸体积分数最低,NaCl质量在1.5 g水平左右时,STC提取率最高。上述现象可解释为酸性条件不利于STC提取,而适当的盐度增强了离子效应,促进盐析分层有利于提高STC提取率。由图5b可知,当甲酸和PSA均处于最低水平时提取率曲面达到最高点,即在本实验中不加入甲酸和PSA效果更好。图5c结果显示,NaCl和无水CH3COONa两种盐的添加量均与STC提取率呈正相关。
在得到的二次回归方程进行响应面分析后,为获取最高的STC提取率,利用软件的Numerical Solutions功能求拟合方程中的响应值Y的最大值,分别对各变量求一阶偏导,计算得到最佳条件为:0%甲酸、1.43 g NaCl、0.95 g无水CH3COONa和0 mg PSA。
2.5 方法学验证结果
2.5.1 基质效应与线性
本实验选取5 份不同的空白鸡蛋进行测试,结果显示:方法平均绝对基质效应为83.6%,RSD为3.8%。尽管绝对基质效应和相对基质效应均处于可接受范围,后续实验仍采用基质匹配曲线法来补偿基质效应以获得更加准确的测定结果,线性范围在0.125~1 000 ng/mL之间,所得线性相关系数为0.999 6。
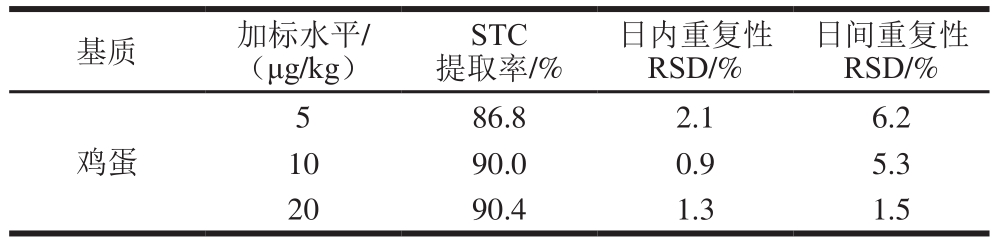
2.5.2 准确性、精密度与方法检测限
准确性及精密度结果如表6所示,该前处理方法测定鸡蛋中STC的检出限为0.1 μg/kg,定量限为0.5 μg/kg。
表6 准确性及精密度结果
Table 6 Accuracy and precision of the method
2.5.3 方法应用与实际样品测定结果
在最佳前处理条件下对45 份鸡蛋样品中的STC进行测定,其中共10 份样品检测结果为阳性(典型空白样品与阳性样品如图6所示),污染含量范围在0.5~3 608 μg/kg(部分样品检测结果见表7),推测可能与母鸡饲养方式及饲养环境有关。所有阳性样品均使用免疫亲和柱法进行复测,将所得2 种方法测定结果进行对比发现并未存在显著性差异(P=0.941>0.05),说明本实验提出的方法足够准确,无明显系统误差。
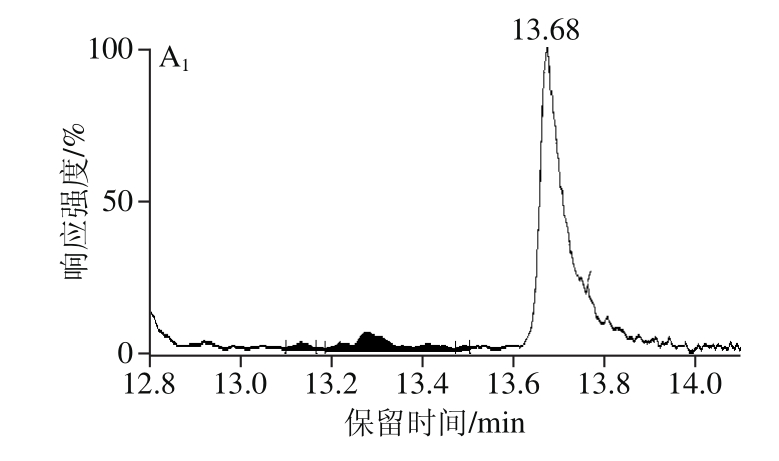
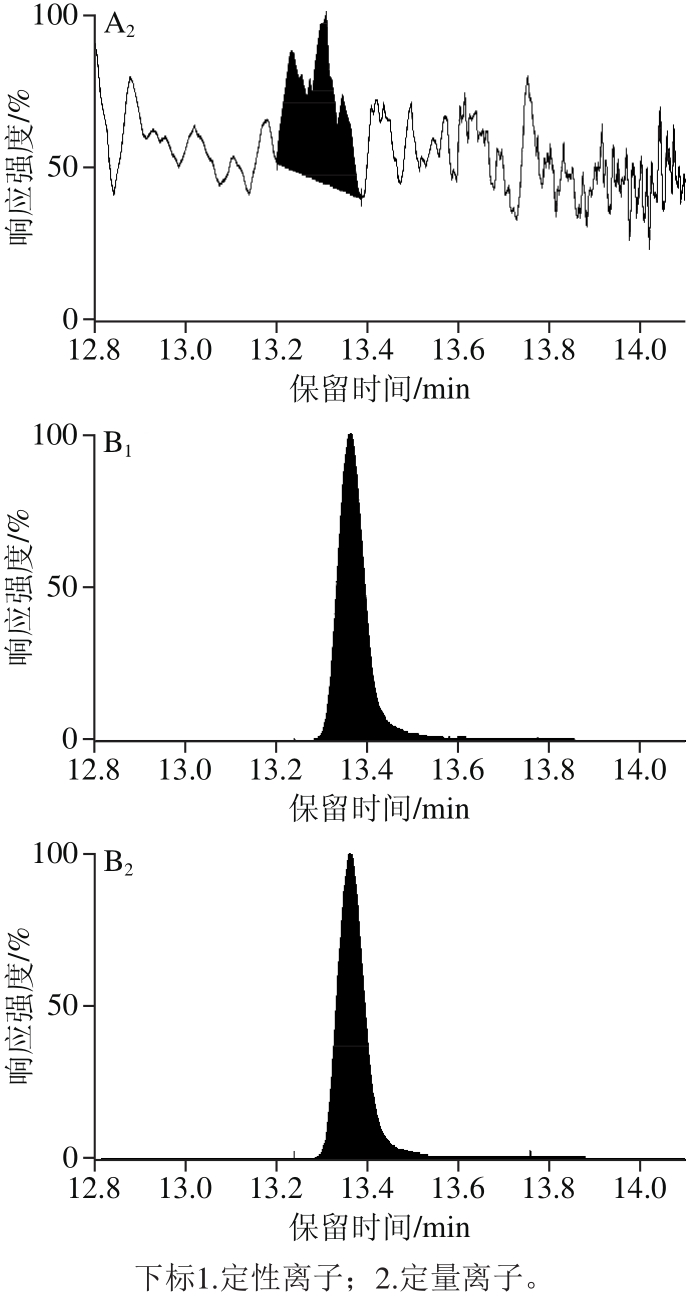
图6 空白鸡蛋样品(A)与阳性鸡蛋样品(B)多反应监测色谱图
Fig. 6 Multiple-reaction monitoring (MRM) chromatograms of blank egg sample (A) and positive egg sample (B)
表7 2 种方法的实际样品测定结果对比
Table 7 Comparison of two methods for the determination of sterigmatocystin in real samples

注:#.测定值高于检出限低于定量限;ND.未检出。
本研究建立了QuEChERS法提取净化鸡蛋中STC的分析方法,实验条件经Plackett-Burman设计筛选后使用中心组合设计试验优化。方法前处理速度快,无需复杂净化手段,待测毒素提取率高,定量结果准确,灵敏度和精密度较好。将阳性样品测定结果与GB 5009.25—2016方法进行对比,显示在2 种方法之间不存在显著性差异。鉴于本研究发现鸡蛋样品中较高的STC污染率,且有高污染浓度样本被检出,建议在适当范围内扩大对鸡蛋中STC的监测,并以此为基础开展STC污染源以及体内代谢机理的研究调查,为食品中STC风险评估工作的开展提供一定参考依据。
参考文献:
[1] RYCHLIK M, LEPPER H, WEIDNER C H, et al. Risk evaluation of the Alternaria mycotoxin tenuazonic acid in foods for adults and infants and subsequent risk management[J]. Food Control, 2016, 68:181-185. DOI:10.1016/j.foodcont.2016.03.035.
[2] MALLY A, SOLFRIZZO M, DEGEN G H. Biomonitoring of the mycotoxin Zearalenone: current state-of-the art and application to human exposure assessment[J]. Archives of Toxicology, 2016, 90(6):1281-1292. DOI:10.1007/s00204-016-1704-0.
[3] ALSHANNAQ A, YU J H. Occurrence, toxicity, and analysis of major mycotoxins in food[J]. International Journal of Environmental Research and Public Health, 2017, 14(6): 632-652. DOI:10.3390/ijerph14060632.
[4] JESTOI M, ROKKA M, JÄRVENPÄÄ E, et al. Determination of Fusarium mycotoxins beauvericin and enniatins (A, A1, B, B1)in eggs of laying hens using liquid chromatography-tandem mass spectrometry (LC-MS/MS)[J]. Food Chemistry, 2009, 115(3): 1120-1127. DOI:10.1016/j.foodchem.2008.12.105.
[5] TANGNI E K, WAEGENEERS N, VAN OVERMEIRE I, et al.Mycotoxin analyses in some home produced eggs in Belgium reveal small contribution to the total daily intake[J]. The Science of the Total Environment, 2009, 407(15): 4411-4418. DOI:10.1016/j.scitotenv.2008.10.060.
[6] 朱闰月, 赵志勇, 杨宪立, 等. 基质固相分散-液相色谱串联质谱法同时检测鸡蛋中15 种真菌毒素生物标志物[J]. 分析化学, 2015, 43(7):994-1000.
[7] YOGENDRARAJAH P, JACXSENS L, LACHAT C, et al. Public health risk associated with the co-occurrence of mycotoxins in spices consumed in Sri Lanka[J]. Food and Chemical Toxicology, 2014, 74:240-248. DOI:10.1016/j.fct.2014.10.007.
[8] ZHENG R, XU H, WANG W, et al. Simultaneous determination of aflatoxin B(1), B(2), G(1), G(2), ochratoxin A, and sterigmatocystin in traditional Chinese medicines by LC-MS-MS[J]. Analytical and Bioanalytical Chemistry, 2014, 406(13): 3031-3039. DOI:10.1007/s00216-014-7750-7.
[9] TURNER P C, BURLEY V J, ROTHWELL J A, et al.Deoxynivalenol: rationale for development and application of a urinary biomarker[J]. Food Additives & Contaminants, 2008, 25(7): 864-871.DOI:10.1080/02652030801895040.
[10] ZHU R, ZHAO Z, WANG J, et al. A simple sample pretreatment method for multi-mycotoxin determination in eggs by liquid chromatography tandem mass spectrometry[J]. Journal of Chromatography A, 2015, 1417:1-7. DOI:10.1016/j.chroma.2015.09.028.
[11] BETINA V. Mycotoxins: chemical, biological and environmental aspects[J]. Elsevier, 1989, 395(5): 463-503. DOI:10.7506/spkx1002-6630-201506039.
[12] HE J, LI X Z, ZHOU T. Sample clean-up methods, immunoaffinity chromatography and solid phase extraction, for determination of deoxynivalenol and deepoxy deoxynivalenol in swine serum[J].Mycotoxin Research, 2009, 25(2): 89-94. DOI:10.1007/s12550-009-0013-3.
[13] WEN J, KONG W, WANG J, et al. Simultaneous determination of four aflatoxins and ochratoxin A in ginger and related products by HPLC with fluorescence detection after immunoaffinity column clean-up and postcolumn photochemical derivatization[J]. Journal of Separation Science, 2013, 36(23): 3709-3716. DOI:10.1002/jssc.201300885.
[14] AFZALI D, GHANBARIAN M, MOSTAFAVI A, et al. A novel method for high preconcentration of ultra trace amounts of B(1),B(2), G(1) and G(2) aflatoxins in edible oils by dispersive liquidliquid microextraction after immunoaffinity column clean-up[J].Journal of Chromatography A, 2012, 1247: 35-41. DOI:10.1016/j.chroma.2012.05.051.
[15] REN Y, ZHANG Y, SHAO S, et al. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultraperformance liquid chromatography tandem mass spectrometry[J].Journal of Chromatography A, 2007, 1143(1/2): 48-64. DOI:10.1016/j.chroma.2006.12.064.
[16] WANG R G, SU X O, CHENG F F, et al. Determination of 26 mycotoxins in feedstuffs by multifunctional clean-up column and liquid chromatography-tandem mass spectrometry[J]. Chinese Journal of Analytical Chemistry, 2015, 43(2): 264-270. DOI:10.1016/s1872-2040(15)60807-6.
[17] SUGITA-KONISHI Y, TANAKA T, NAKAJIMA M, et al. The comparison of two clean-up procedures, multifunctional column and immunoaffinity column, for HPLC determination of ochratoxin A in cereals, raisins and green coffee beans[J]. Talanta, 2006, 69(3):650-655. DOI:10.1016/j.talanta.2005.10.036.
[18] ARROYO-MANZANARES N, HUERTAS-PEREZ J F, GAMIZGRACIA L, et al. Simple and efficient methodology to determine mycotoxins in cereal syrups[J]. Food Chemistry, 2015, 177: 274-279.DOI:10.1016/j.foodchem.2015.01.040.
[19] LIU Y, HAN S, LU M, et al. Modified QuEChERS method combined with ultra-high performance liquid chromatography tandem mass spectrometry for the simultaneous determination of 26 mycotoxins in sesame butter[J]. Journal of Chromatography B, 2014, 970: 68-76.DOI:10.1016/j.jchromb.2014.06.033.
[20] XU J J, ZHOU J, HUANG B F, et al. Simultaneous and rapid determination of deoxynivalenol and its acetylate-derivatives in wheat flour and rice by ultra high performance liquid chromatography with photo diode array detection[J]. Journal of Separation Science, 2016,39(11): 2028-2036. DOI:10.1002/jssc.201501316.
[21] LAI X, LIU R, RUAN C, et al. Occurrence of aflatoxins and ochratoxin A in rice samples from six provinces in China[J]. Food Control, 2015, 50: 401-404. DOI:10.1016/j.foodcont.2014.09.029.
[22] ARROYO-MANZANARES N, HUERTAS-PEREZ J F, GAMIZGRACIA L, et al. A new approach in sample treatment combined with UHPLC-MS/MS for the determination of multiclass mycotoxins in edible nuts and seeds[J]. Talanta, 2013, 115: 61-67. DOI:10.1016/j.talanta.2013.04.024.
[23] ZHOU J, XU J J, HUANG B F, et al. High-performance liquid chromatographic determination of multi-mycotoxin in cereals and bean foodstuffs using interference-removal solid-phase extraction combined with optimized dispersive liquid-liquid microextraction[J]. Journal of Separation Science,2017, 40(10): 2141-2150. DOI:10.1002/jssc.201601326.
[24] 曲斌. QuEChERS在动物源性食品兽药残留检测中的研究进展[J].食品科学, 2013, 34(5): 327-331. DOI:10.7506/spkx1002-6630-201305068.
[25] WALORCZYK S. Development of a multi-residue method for the determination of pesticides in cereals and dry animal feed using gas chromatography-tandem quadrupole mass spectrometry Ⅱ. improvement and extension to new analytes[J]. Journal of Chromatography A, 2008,1208(1/2): 202-214. DOI:10.1016/j.chroma.2008.08.068.
[26] FRENICH A G, ROMERO-GONZáLEZ R, GÓMEZ-PéREZ M L,et al. Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry[J]. Journal of Chromatography A, 2011, 1218(28): 4349-4356. DOI:10.1016/j.chroma.2011.05.005.
[27] VISWANATHAN C T, BANSAL S, BOOTH B, et al. Workshop/conference report-quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays[J]. Journal of the American Association of Pharmaceutical Scientists, 2007, 9(1): 30-42.
[28] 湛东锐, 李连强, 曹娜, 等. 盐生海芦笋内生真菌Salicorn 35的分离鉴定与抗氧化发酵条件优[J]. 食品科学, 2013, 34(19): 158-165.DOI:10.7506/spkx1002-6630-201319034.
[29] RANJBARI E, HADJMOHAMMADI M R. Optimization of magnetic stirring assisted dispersive liquid-liquid microextraction of rhodamine B and rhodamine 6G by response surface methodology∶ application in water samples, soft drink, and cosmetic products[J]. Talanta, 2015,139∶ 216-225. DOI∶10.1016/j.talanta.2015.02.051.
[30] ASADOLLAHZADEH M, TAVAKOLI H, TORAB-MOSTAEDI M, et al. Response surface methodology based on central composite design as a chemometric tool for optimization of dispersivesolidification liquid-liquid microextraction for speciation of inorganic arsenic in environmental water samples[J]. Talanta, 2014, 123∶ 25-31.DOI∶10.1016/j.talanta.2013.11.071.
Optimization and Validation of QuEChERS Extraction for Determination of Sterigmatocystin in Eggs by Response Surface Methodology
ZHOU Jian1,2, XU Jiaojiao1,*, JIN Micong2, CAI Zengxuan1, HUANG Baifen1, REN Yiping3
(1. Department of Physicochemical & Toxicology, Zhejiang Provincial Centre for Disease Control and Prevention, Hangzhou 310051, China;2. Zhejiang Provincial Key Lab of Health Risk Appraisal for Trace Toxic Chemicals, Ningbo Municipal Centre for Disease Control and Prevention, Ningbo 315010, China; 3. Zhejiang Yangtze Delta Region Institute of Tsinghua University, Application Technology Cooperation Center of National Center for Food Safety Risk Assessment, Jiaxing 314006, China)
Abstract:A quick, easy, cheap, effective, rugged, and safe (QuEChERS) extraction procedure was presented andoptimized for the determination of sterigmatocystin in eggs by high performance liquid chromatography-mass spectrometry (HPLC-MS).The analyte was extracted from samples with aqueous acetonitrile solution, followed by salting out using anhydrous NaSO4,NaCl and CH3COONa,and then purification and concentration with C18sorbent and anhydrous Na2SO4before analysis.Optimization of experimental conditions for maximum extraction efficiency was carried out using Plackett-Burman design,one-factor-at-a-time method and response surface methodology. The matrix-matched external standard calibration was employed for quantification. Moreover, all positive sampleswere confirmed by immunoaffinity column chromatography. The results of validation showed that good linearity with a correlation coefficient (R2) > 0.999 6 was achieved within the range from 0.125 to 1 000 ng/mL. The limit of detection (LOD) and the limit of quantification (LOQ) for sterigmatocystin in eggs were 0.1 and 0.5 μg/kg, respectively. Satisfactory recovery (86.8%-90.4%) and inter-day reproducibility (RSD, 1.5%-6.2%)were obtained with blank egg matrices at three spiked levels. Finally, the established method was applied to analyze 45 real samples, 10 of which were positive at concentrations of 0.5 to 3 608 μg/kg.
Key words:egg; sterigmatocystin; QuEChERS extraction; response surface methodology; high performance liquid chromatography-mass spectrometry
DOI:10.7506/spkx1002-6630-201724047
中图分类号:O657.63
文献标志码:A
文章编号:1002-6630(2017)24-0288-08
收稿日期:2017-03-02
作者简介:周健(1991—),男,硕士,主要从事真菌毒素研究。E-mail:1428541555@qq.com
*通信作者:许娇娇(1987—),女,硕士,主要从事食品安全检验技术研究。E-mail:jjxucdc@163.com
引文格式:
周健, 许娇娇, 金米聪, 等. 响应面试验优化QuEChERS法提取鸡蛋中杂色曲霉毒素工艺及方法学验证[J]. 食品科学,2017, 38(24): 288-295.
DOI:10.7506/spkx1002-6630-201724047. http://www.spkx.net.cn
ZHOU Jian, XU Jiaojiao, JIN Micong, et al. Optimization and validation of QuEChERS extraction for determination of sterigmatocystin in eggs by response surface methodology[J]. Food Science, 2017, 38(24)∶ 288-295. (in Chinese with English abstract) DOI∶10.7506/spkx1002-6630-201724047. http∶//www.spkx.net.cn