表1 本研究中使用的菌株与质粒
Table1 Strains and plasmids used in this research

?
陆 浩 1,李天明 1,刘金雷 1,杜红燕 1,王北辰 2,冯惠勇 1,*
(1.河北科技大学生物科学与工程学院,河北 石家庄 050018;2.威斯康星大学农业与生命学院,威斯康星 麦迪逊 53706)
摘 要:通过构建尿嘧啶磷酸核糖转移酶基因(upp)敲除打靶质粒,采用同源重组的方法获得了生酮古龙酸杆菌(Ketogulonigenium vulgare)的upp基因缺失突变株K. vulgare Δupp,可作为基因组无痕修饰系统底盘细胞的upp基因表达载体,转化突变株K. vulgare Δupp,获得upp基因回补菌株PBB-upp,并验证了upp作为负向筛选标记的可行性。实验结果表明:突变株Δupp在1 mg/mL的5-氟尿嘧啶培养基上可生长,野生型和回补菌株PBB-upp不能生长,3株菌对5-氟尿嘧啶的抗性存在显著差异,说明可以将upp基因作为负向筛选标记,与正向筛选标记相结合,在upp基因缺失的底盘细胞基础上通过两次同源重组,实现基因组的无痕修饰与改造。
关键词:生酮古龙酸杆菌;负向筛选标记;无痕改造;5-氟尿嘧啶
VC又名L-抗坏血酸,是人体必需的水溶性维生素之一。“两步发酵法”是目前VC的主要工业化生产方式 [1-3]。生酮古龙酸杆菌(Ketogulonigenium vulgare)是目前VC两步发酵工艺的重要生产菌,主要与芽孢杆菌等伴生菌完成糖酸转化 [4]。优良的菌种对于提高VC产率和经济效益具有重要意义。
随着分子生物学的发展及K. vulgare全基因组的测序完成 [5-6],利用代谢工程技术进行菌种改造正成为VC生产菌育种的主要手段。目前采用的基因敲除、基因敲入及点突变修饰等基因组改造技术中,多采用的是抗性筛选标记,存在一些弊端:被改造菌株可被使用的抗生素数量有限;多基因改造会因抗性标记的叠加使用影响目的菌的正常生长;抗性基因本身是外源基因,并不是宿主菌生长所必需,在食品行业产品使用中受到限制等 [7-9]。所以,迫切需要研发K. vulgare基因组的无痕改造技术,以克服传统的基因组改造技术中“疤痕”的存在和筛选标记不能反复使用的不足,实现多个基因的连续敲除,最终获得VC高产优良菌种。
K. vulgare中存在upp基因,可编码尿嘧啶磷酸核糖转移酶(uracil phosphoribosyl transferase,UPRT),使细胞不能合成尿嘧啶,只能利用胞外尿嘧啶。5-氟尿嘧啶是胸腺嘧啶类似物,也能作为UPRT的底物被转化成胸苷酸合成酶的强烈抑制剂,即5-氟单磷酸脱氧尿嘧啶(5-f l uoro-2’-deoxyuridine-5’-monophosphate,5-F-dUMP),5-F-d U M P的存在会导致宿主细胞死亡 [1 0-11]。而upp基因缺失后菌体不能利用5-氟尿嘧啶产生毒性,可在含5-氟尿嘧啶的培养基中生长,从而使第2次重组后失去抗性基因和upp基因的突变株得以筛选。因此,借助5-氟尿嘧啶的毒性,UPRT可作为负向筛选标记使用。本实验利用同源重组,敲除了染色体上UPRT的全长基因upp,构建得到底盘细胞K. vulgare Δupp突变株,经过upp基因回补实验,验证了UPRT作为负向筛选标记使用的可行性。在此底盘细胞的基础上可以将upp基因作为负向筛选标记,进行基因的无痕敲入、敲除及修饰,克服了使用抗生素筛选标记的缺陷,为实现K. vulgare基因组无痕修饰,获得具有高产优良性状的优势遗传资源提供理论和技术支持。
1.1 材料与试剂
1.1.1 菌株和载体
表1 本研究中使用的菌株与质粒
Table1 Strains and plasmids used in this research
?
1.1.2 培养基
K. vulgare培养基(含卡那霉素):葡萄糖20.0 g/L、玉米浆10.0 g/L、KH 2PO 41.0 g/L、硫酸铵4.0 g/L、硫酸镁0.2 g/L、碳酸钙0.1 g/L、卡那霉素50 μg/mL、琼脂20.0 g/L,pH 6.4~6.7。
LB培养基:胰蛋白胨10.0 g/L、酵母提取物5.0 g/L、氯化钠10.0 g/L,pH 7.0。
K. vulgare固体培养基:山梨醇20.0 g/L、玉米浆5.0 g/L、磷酸二氢钾0.5 g/L、磷酸二氢铵0.5 g/L、硫酸铵2.0 g/L、硫酸镁0.2 g/L、碳酸钙0.1 g/L、琼脂20.0 g/L,pH 6.4~6.7。
K. vulgare筛选培养基:蔗糖100.0 g/L、山梨醇20.0 g/L、玉米浆5.0 g/L、KH 2PO 40.5 g/L、磷酸二氢铵0.5 g/L、硫酸铵2.0 g/L、硫酸镁0.2 g/L、碳酸钙0.1 g/L、琼脂20.0 g/L,pH 6.4~6.7。
1.1.3 试剂
ExTaq聚合酶、限制性内切酶EcoRⅠ和BamHⅠ宝生物工程(大连)有限公司;HiFi DNA聚合酶北京全式金生物技术有限公司;T4 DNA连接酶 NEB(北京)有限公司;氨苄青霉素、卡那霉素 上海生工生物工程有限公司;5-溴-4-氯-3-吲哚-β-D-半乳糖苷(5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside,X-gal) 美国Sigma公司;异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-1-thiogalactopyranoside,IPTG) 鼎国生物工程公司;5-氟尿嘧啶 上海谱振生物科技有限公司;DNA回收试剂盒、质粒提取试剂盒 天根生化科技(北京)有限公司。
1.2 仪器与设备
聚合酶链式反应(polymerase chain reaction,PCR)仪、台式高速离心机 德国Eppendorf公司;全自动凝胶成像系统 美国Bio-Rad公司;高速冷冻离心机 美国Thermo公司;酶标仪 美国BioTek公司。
1.3 方法
1.3.1 K. vulgare对5-氟尿嘧啶的敏感性测定
5-氟尿嘧啶对K. vulgare野生型有一定毒性,会抑制其生长,当5-氟尿嘧啶质量浓度达到一定值时会使细胞死亡;为了确定其对K. vulgare生长抑制的最低质量浓度,在含不同质量浓度的5-氟尿嘧啶液体培养基和固体培养基中对野生型K. vulgare进行培养。根据文献[12]设定测试的5-氟尿嘧啶最高质量浓度为2 mg/mL,随后通过二倍稀释法逐步降低5-氟尿嘧啶的质量浓度,选用4 个质量浓度梯度,分别为0、0.5、1.0、2.0 mg/mL。为保证接种量一致,接种前用无菌0.9% NaCl溶液稀释调整菌悬液的OD 600 nm为0.7,4 个5-氟尿嘧啶质量浓度各接种菌液10 μL。液体培养基放置在30 ℃条件下,180 r/min摇床中振荡培养,每隔6 h观察记录菌株生长情况。固体平板用相同的接种环接种后放置在30 ℃恒温培养箱中,倒置培养,24 h后观察记录菌株生长情况。
1.3.2 敲除upp基因打靶载体的构建
以K. vulgare基因组DNA、pEASY质粒为模板,利用PCR扩增出upp基因的上下游同源臂和卡那抗性基因片段,将3 个片段等物质的量比例混合后作为模板,通过重叠PCR获得目的条带,PCR产物纯化后与pMD18-T载体相连,转化进大肠杆菌Trans 5α感受态细胞内,经菌落PCR验证,获得打靶载体pMD18-T-xqupp,送由上海生工股份有限公司测序。
1.3.3 K. vulgare Δupp菌株的构建
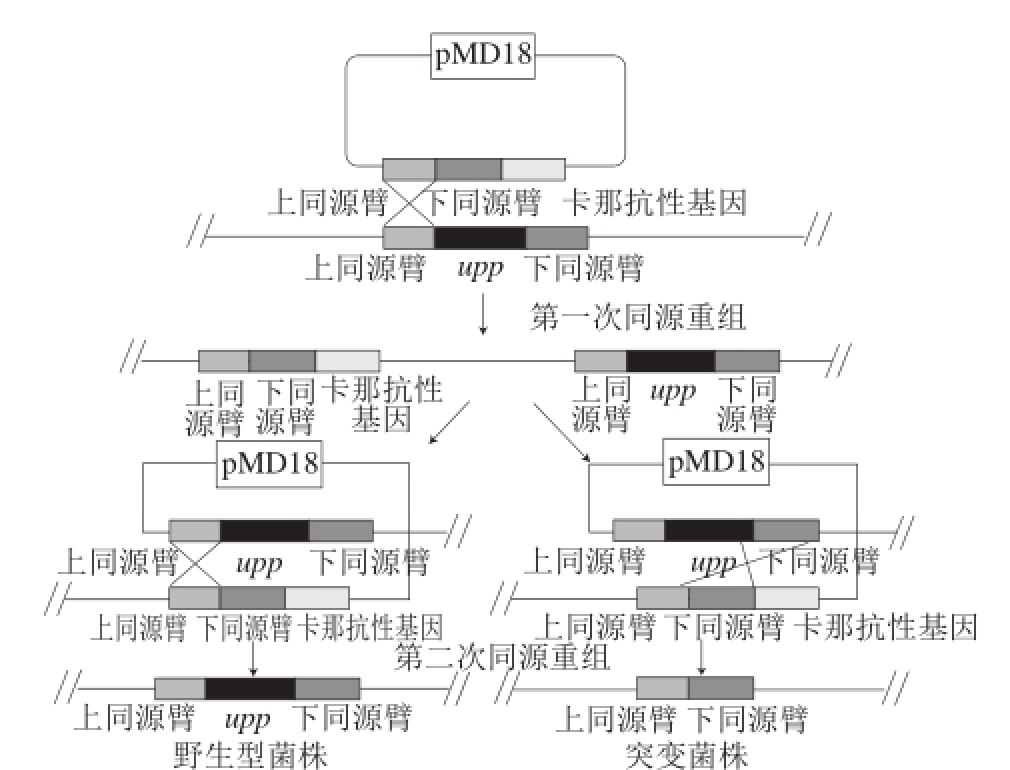
图1 利用同源双交换进行基因敲除原理示意图
Fig.1 Schematic diagram of gene deletion based on homologous double switch
K. vulgare中upp基因的敲除是通过2 次同源重组来实现的(图1)。将重组质粒pMD18-T-xqupp电击转化至野生型K. vulgare感受态细胞中,通过第1次同源重组卡那霉素抗性基因被整合到染色体上,利用卡那霉素固体平板筛选出发生重组的菌株,将发生第1次重组的菌株在液体培养基中培养后会自发产生第2次同源重组,此时可能出现upp基因缺失菌株和缺失基因恢复菌株,将菌液稀释涂布于含5-氟尿嘧啶的固体平板上,长出转化子后分别挑取后转接至含5-氟尿嘧啶和卡那霉素的平板上,5-氟尿嘧啶培养基上生长而卡那霉素培养基上不能生长的菌株则为目的菌株。
将得到的pMD18-T-xqupp打靶载体通过电转化(电击条件为:2.5 kV/cm电压、250 Ω电阻、25 μF电容、2 mm电击杯)于K. vulgare野生型感受态细胞中,30 ℃条件下培养48 h,挑取转化子,利用菌落PCR对转化子进行验证,从而获得upp基因缺失菌株K. vulgare Δupp。
1.3.4 upp基因回补菌株的构建
以K. vulgare基因组为模板,利用PCR扩增出upp基因,将PCR片段回收后与空质粒pBBRIMCS-5分别用EcoRⅠ和BamHⅠ双酶切,回收目的片段后通过T4连接酶16 ℃连接过夜,转化至大肠杆菌Trans 5α感受态细胞,利用菌落PCR验证出构建成功的upp基因回补质粒pBBR1MCS-5-upp。将构建成功的回补质粒pBBR1MCS-5-upp电转化到已获得的upp基因缺失菌株K. vulgare Δupp的感受态细胞中,通过PCR验证获得upp基因回补菌株K. vulgare Δupp/upp。
1.3.5 工程菌生理活性验证
利用1.3.1节方法,分别在含有不同质量浓度5-氟尿嘧啶的液体培养基和固体培养基中培养upp基因缺失菌株K. vulgare Δupp和upp基因回补菌株K. vulgare Δupp/upp,定时观测记录菌株生长情况,研究工程菌株对5-氟尿嘧啶的耐受性。
2.1 K. vulgare对5-氟尿嘧啶的敏感性分析
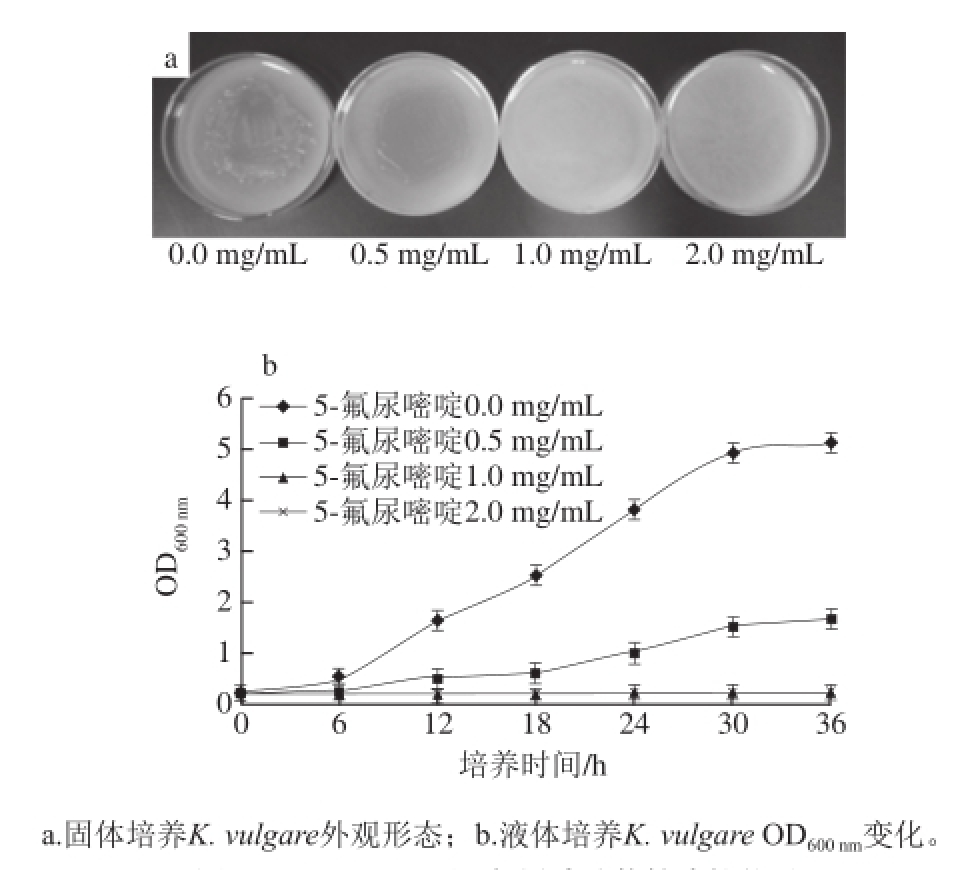
图2 K. vulgare对5-氟尿嘧啶的敏感性结果
Fig.2 5-Fluorouracil sensitivity of K. vulgare cultivated in solid (a) and liquid (b) media
如图2所示,当5-氟尿嘧啶质量浓度为1 mg/mL时,固体培养基和液体培养基中培养的野生型K. vulgare不能正常生长,因此确定5-氟尿嘧啶对K. vulgare生长起到抑制作用的最低质量浓度为1 mg/mL。
2.2 upp基因缺失菌株的获得
2.2.1 打靶载体的构建及验证
通过融合PCR技术,构建带有upp基因的上下游同源臂和卡那抗性基因片段的重组质粒pMD18-T-xqupp。经PCR验证,条带与设计条带大小一致,结果如图3。
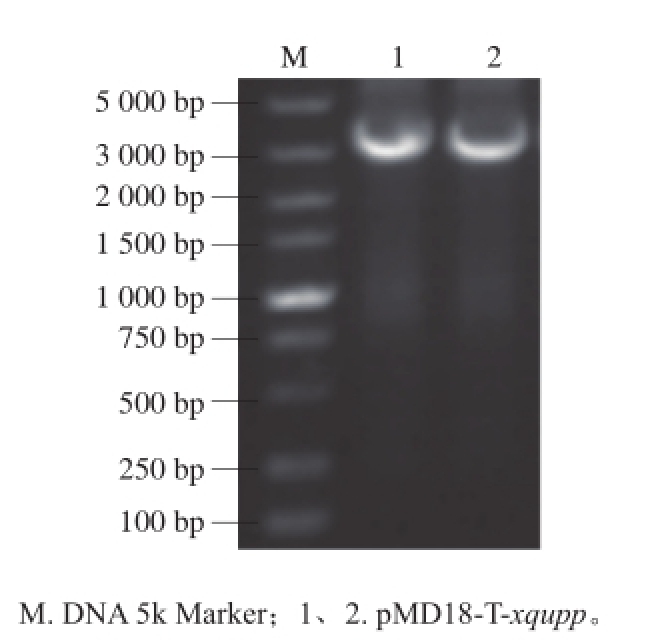
图3 pMD18-T-xqupp质粒PCR验证
Fig.3 PCR identifi cation of plasmid pMD18-T-xqupp
2.2.2 upp基因缺失菌株的筛选
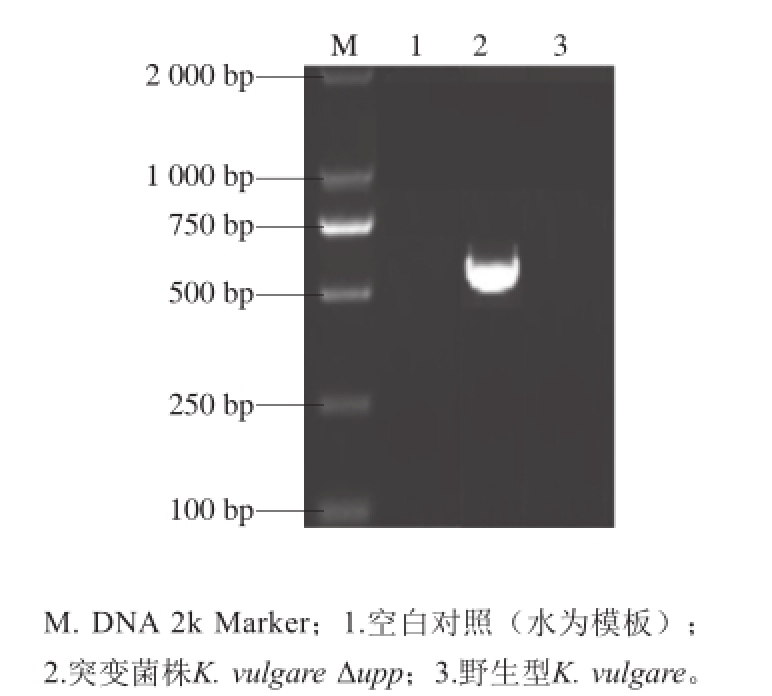
图4 K. vulgareΔupp突变株菌落PCR验证
Fig.4 Validation of K. vulgare Δ upp deletion mutant
将打靶质粒转化K. vulgare野生型感受态细胞,得到具有卡那抗性的阳性转化子,利用菌落PCR扩增upp基因,结果如图4所示,野生型K. vulgare在目的位置能够扩增出条带,而工程菌株在目的位置没有条带,从而获得了upp基因缺失的工程菌K. vulgare Δupp。
2.3 upp基因回补菌株的构建
构建upp基因回补质粒pBBR1MCS-5-upp,转化大肠杆菌Trans 5α,经酶切验证,如图5B所示,质粒大小4 800 bp,片段大小630 bp,与理论值一致,说明质粒构建成功。将质粒pBBR1MCS-5-upp经电转化至K. vulgare Δupp菌株中,得到转化子K. vulgare Δupp/upp,分别对upp基因缺失菌株K. vulgare Δupp和转化子K. vulgare Δupp/upp进行PCR验证,如图5C所示,菌株K. vulgare Δupp不能扩增出upp基因,而K. vulgare Δupp/upp能够扩增upp基因,说明K. vulgare Δupp/upp菌株中回补了upp基因。
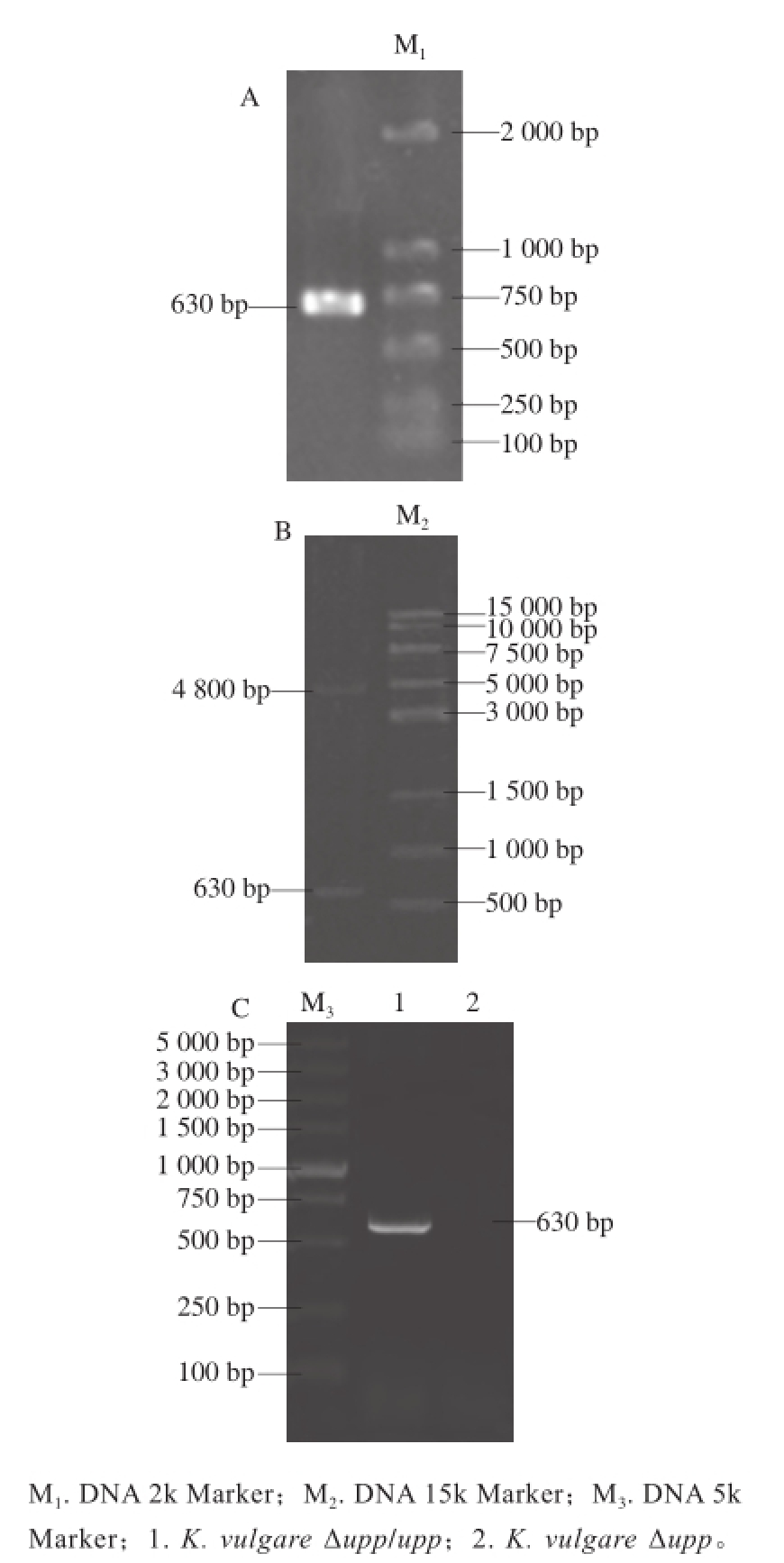
图5 upp 基因的PCR扩增(A)、pBBR1MCS-5-upp 质粒酶切(B)及的鉴定(C)
Fig.5 PCR Amplif i cation of upp gene (A), enzyme digestion identif i cation of pBBR1MCS-5-upp plasmid (B) and identif i cation of K. vulgare Δupp/upp (C) K. vulgareΔupp/upp
2.4 工程菌株生理活性验证
根据UPRT的催化功能和5-氟尿嘧啶的毒性原理,upp基因缺失菌株能在含5-氟尿嘧啶的筛选培养基上生长,而野生型菌株和回补upp基因的工程菌株不能够生长。故将K. vulgare、K. vulgare Δupp和K. vulgare Δupp/upp分别接种在含0、1 mg/mL的5-氟尿嘧啶固体和液体培养基中培养,观察菌体的生长情况,结果如图6。验证结果表明upp基因缺失菌株K. vulgare Δupp在含5-氟尿嘧啶的培养基上能够正常生长,而upp基因回补菌株K. vulgare Δupp/upp与野生型菌株K. vulgare一样,均在不能在含1 mg/mL 5-氟尿嘧啶的培养基上生长,这3 株菌对5-氟尿嘧啶的抗性存在显著差异,说明可以将upp基因作为负向筛选标记,与正向筛选标记相结合,在upp基因缺失底盘细胞基础上通过两次同源重组,实现基因组的无痕修饰与改造。
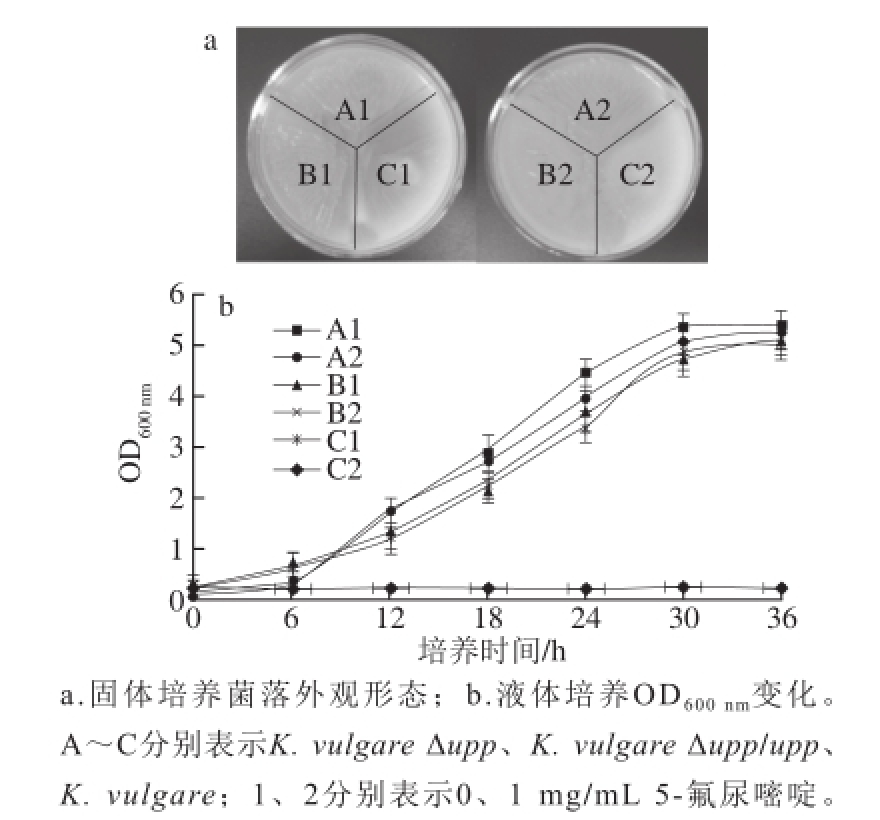
图6 5-氟尿嘧啶对基因工程菌生长状况的影响
Fig.6 Effects of 5-f l uorouracil of the growth of mutant strains
利用负向筛选标记实现基因组无痕修饰改造方面的研究已取得一定成果。例如Karlinsey [13]于2007年利用Bochner-Maloy培养基与抗性筛选标记tetAR相结合,完成对沙门氏菌(Salmonella)的基因无痕敲除。后Cox [14]、Gerlach [15]等也均采用无痕敲除方法完成对沙门菌基因组的无痕修饰改造。Mizoguchi [16]、Yang Junjie [17]等选用sacB基因作为负向筛选标记,成功构建大肠杆菌(Escherichia coli)的基因无痕敲除、插入系统。2011年刘霞等 [18]基于同源重组技术用sacB重组载体成功敲除副溶血弧菌(Vibrio parahaemolyticus)的opaR基因。同年Kato等 [19]实现了sarS基因在金黄色葡萄球菌(Staphylococcus aureus)的无痕敲除。但对于K. vulgare的此类研究还处于空白。
UPRT广泛存在于原核生物、某些低等真核生物细胞中,其作为负向筛选标记还鲜见相关报道。K. vulgare中的upp基因全长630 bp,编码尿嘧啶生成UPRT,使细胞合成尿嘧啶。5-氟尿嘧啶是胸腺嘧啶类似物也能作为UPRT的底物被转化成胸苷酸合成酶的强烈抑制剂,即5-F-dUMP,5-F-dUMP的存在会抑制胸腺嘧啶合成酶活性,导致宿主细胞死亡。利用这一特性,尿嘧啶生成UPRT的表达与否,直接影响菌株对5-氟尿嘧啶的抗性,影响菌株的存活与否。利用这种生长差异,借助5-氟尿嘧啶的筛选培养基,就可以对重组菌进行筛选。
本实验通过重叠PCR [20-21]、TA克隆 [22]、转化 [23-25]等方法获得打靶质粒。制备宿主菌感受态细胞,将打靶载体电转至宿主菌感受态中,经2 次同源重组筛选出了K. vulgare Δupp突变株,生理验证该菌株能在含1 mg/mL 5-氟尿嘧啶培养基上生长,本实验获得的upp基因缺失突变株及其对5-氟尿嘧啶的抗性性状研究,对于无痕遗传修饰技术发展具有重要的参考价值。
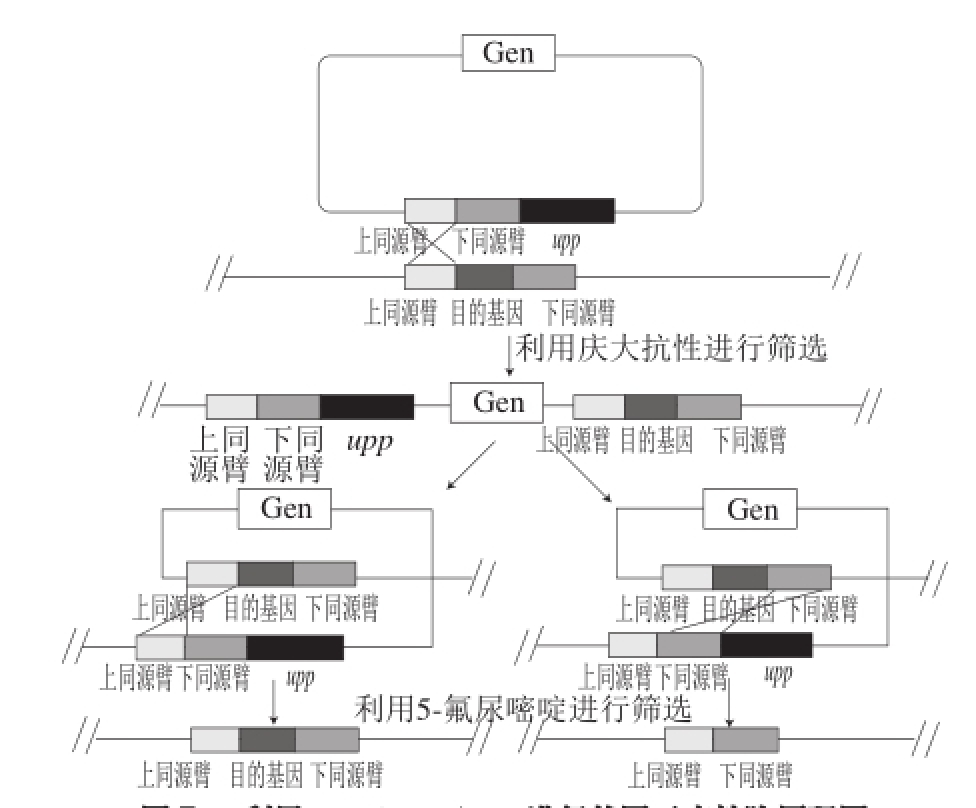
图7 利用K. vulgareΔupp进行基因无痕敲除原理图
Fig.7 Schematic of scarless gene knockout based on K. vulgare Δupp
为了验证upp基因作为负向筛选标记的可行性,本实验在upp缺失菌株基础上,回补了upp基因,该菌株在1 mg/mL 5-氟尿嘧啶培养基上不生长,说明可实现在upp缺失的底盘细胞中利用一次交换2 次重组进行基因无痕敲除(图7)。打靶质粒的构建包括被敲除基因的上游同源臂、抗性筛选标记基因、可表达的upp基因和被敲除基因的下游同源臂,第1次重组通过抗性筛选后,将得到的转化子在无抗生素的培养基上孵育后,再涂到含有5-氟尿嘧啶的筛选平板上,只有实现二次重组的转化子,由于去除了抗性基因和upp基因得以存活,得到的阳性转化子既敲除了目标基因又没有抗性基因残留,以此建立了K. vulgare基因组无痕修饰改造系统,这在国内外属先进技术。本研究结果不仅为代谢工程改造K. vulgare获得优良VC高产菌株提供了理论和技术支持,也为其他工业菌株的代谢工程改造提供了技术方法的借鉴。
参考文献:
[1] 周彬, 李怡, 刘耀平, 等. VC二步发酵中的微生物生态调控[J].应用生态学报, 2002, 13(11): 1452-1454. DOI:10.13287/j.1001-9332.2002.0336.
[2] 吕淑霞, 冯树, 张忠泽, 等. VC二步发酵中伴生菌的作用机制[J].微生物学报, 2001, 28(5): 10-13. DOI:10.13344/j.microbiol. china.2001.05.003.
[3] 王虹. 维生素C两步发酵中关键步骤的分子生物学研究[D]. 北京:中国人民解放军军事医学科学院, 2005: 5-9.
[4] YANG F, JIA Q, XIONG Z, et al. Complete genome analysis of Ketogulonigeium sp. WB0104[J]. Chinese Science Bulletin, 2006, 51(8): 941-945. DOI:10.1007/s11434-006-0941-7.
[5] XIONG X, HAN S, WANG J, et al. Complete genome sequence of the bacterium Ketogulonicigenium vulgare Y25[J]. Journal of Bacteriology, 2011, 193(1): 315-316. DOI:10.1128/JB.01189-10.
[6] ZOU W, LIU L, ZHANG J, et al. Reconstruction and analysis of a genome-scale metabolic model of the vitamin C producing industrial strain Ketogulonicigenium vulgare WSH-001[J]. Journal of Biotechnology, 2012, 161(1): 42-48. DOI:10.1016/ j.jbiotec.2012.05.015.
[7] 杨奇. 大肠杆菌DH5α upp基因的敲除及其应用研究[D]. 南京: 南京理工大学, 2013: 4-10.
[8] BAILEY J E. Toward a science of metabolic engineering[J]. Science, 1991, 252: 1668-1675. DOI:10.1126/science.2047876.
[9] 胡逢雪, 丁锐, 崔震海, 等. 大肠杆菌基因无痕敲除技术及策略[J].生物技术通讯, 2013, 24(4): 552-557.
[10] HASEGAWA N, ABEIL M, YOKOYAMA K K, et al. Cyclophosphamide enhances antitumor efficacy of oncolytic adenovirus expressing uracil phosphoribosyltransferase (UPRT) in immunocompetent Syrian hamsters[J]. International Journal of Cancer, 2013, 133: 1479-1488. DOI:10.1002/ijc.28132.
[11] ANDERSEN P S, SMITH J M, MYGIND B. Characterization of the upp gene encoding uracil phosphoribosyltransferase of Escherichia coli K12[J]. Federation of European Biochemical Societies, 1992, 204(1): 51-56. DOI:10.1111/j.1432-1033.1992.tb16604.x.
[12] ISLAM S, HASSAN F, TUMURKHUU G, et al. 5-Fluorouracil prevents lipopolysaccharide-induced nitric oxide production in RAW 264.7 macrophage cells by inhibiting Akt-dependent nuclear factorkappa B activation[J]. Cancer Chemotherapy and Pharmacology, 2007, 59(2): 227-233. DOI:10.1007/s00280-006-0261-2.
[13] KARLINSEY J E. lambda-Red genetic engineering in Salmonella enterica serovar Typhimurium[J]. Methods in Enzymology, 2007, 421: 199-209. DOI:10.1016/S0076-6879(06)21016-4.
[14] COX M M, LAYTON S L, JIANG T S, et al. Scarless and sitedirected mutagenesis in Salmonella enteritidis chromosome[J]. BMC Biotechnology, 2007, 7: 59. DOI:10.1186/1472-6750-7-59.
[15] GERLACH R G, JÄCKEL D, HÖLZER S U, et al. Rapid oligonucleotide-based recombineering of the chromosome of Salmonella enterica[J]. Applied and Environmental Microbiology, 2009, 75(6): 1575-1580. DOI:10.1128/AEM.02509-08.
[16] MIZOGUCHI H, TANAKA-MASUDA K, MORI H. A simple method for multiple modif i cation of the Escherichia coli K-12 chromosome[J]. Bioscience, Biotechnology, and Biochemistry, 2007, 71(12): 2905-2911. DOI:10.1271/bbb.70274.
[17] YANG J J, SUN B B, HUANG H, et al. High-efficiency scarless genetic modification in Escherichia coli by using lambda-red recombination and I-SceI cleavage[J]. Applied and Environmental Microbiology, 2014, 80(13): 3826-3834. DOI:10.1128/AEM.00313-14.
[18] 刘霞, 高鹤, 杨琳, 等. 副溶血性弧菌基因敲除方法的建立及应用[J].中国实验动物学报, 2011, 19(3): 188-192.
[19] KATO F, SUGAI M. A simple method of markerless gene deletion in Staphylococcus aureus[J]. Journal of Microbiological Methods, 2011, 87(1): 76-81. DOI:10.1016/j.mimet.2011.07.010.
[20] URBAN A, NEUKIRCHEN S, JAEGER K E. A rapid and efficient method for site-directed mutagenesis using one-step overlap extension PCR[J]. Nucleic Acids Research, 1997, 25(11): 2227-2228. DOI:10.1093/nar/25.11.2227.
[21] 徐芳, 姚泉洪, 熊爱生, 等. 重叠延伸PCR技术及其在基因工程上的应用[J]. 分子植物育种, 2006, 4(5): 747-750.
[22] HOLTON T A, GRAHAM M W. A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors[J]. Nucleic Acids Research, 1991, 19(5): 1156. DOI:10.1093/nar/19.5.1156.
[23] DOWER W J, MILLER J F, RAGSDALE C W. High efficiency transformation of E. coli by high voltage electroporation[J]. Nucleic Acids Research, 1988, 16(13): 6127-6145. DOI:10.1093/ nar/16.13.6127.
[24] MANDEL M, HIGA A. Calcium-dependent bacteriophage DNA infection[J]. Journal of Molecular Biology, 1970, 53(1): 159-162. DOI:10.1016/0022-2836(70)90051-3.
[25] COHEN S N, CHANG A C Y, HSU L. Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA[J]. Proceedings of the National Academy of Sciences of the United States of America, 1972, 69(8): 2110-2114.
Construction of a Marker-Free Deletion System Based on the Uracil Phosphoribosyl Transferase Gene as a Negative Selection Marker in Ketogulonigenium vulgare
LU Hao
1, LI Tianming
1, LIU Jinlei
1, DU Hongyan
1, WANG Beichen
2, FENG Huiyong
1,*
(1. College of Bioscience & Bioengineering, Hebei University of Science and Technology, Shijiazhuang 050018, China; 2. College of Agriculture and Life Science, University of Wisconsin, Madison 53706, USA)
Abstract:By constructing the targeting vector of the uracil phosphoribosyl transferase gene (upp), the upp gene deletion mutant K. vulgare Δupp was obtained using homologous recombination. The mutant was selected as the chassis cell in seamlesss genome modification system. An upp gene expression vector was constructed and transformed into mutant K. vulgare Δupp. After the transformation, an upp gene reverse mutation strain named PBB-upp was obtained, and the feasibility of upp as a negative selectable marker was verified. The results showed that Δupp mutants rather than the wildtype and reverse PBB-upp strains could grow in a medium with 1 mg/mL 5-fluorouracil. Moreover, there were signif i cant differences among the three strains in terms of 5-f l uorouracil resistance, indicating that by combining the upp gene as a negative marker with a positive marker, double homologous recombinations could occur on the chassis cell without the upp gene. In this way, the modif i cation can be achieved without antibiotic marker in Ketogulonigenium vulgare.
Key words:Ketogulonigenium vulgare; negative selection marker; scarless gene alteration; 5-f l uorouracil
DOI:10.7506/spkx1002-6630-201704007
中图分类号:Q789
文献标志码:A
文章编号:1002-6630(2017)04-0039-06
引文格式:
陆浩, 李天明, 刘金雷, 等. 基于尿嘧啶磷酸核糖转移酶为负向筛选标记的生酮古龙酸杆菌基因组无痕修饰系统的构建[J].食品科学, 2017, 38(4): 39-44. DOI:10.7506/spkx1002-6630-201704007. http://www.spkx.net.cn
LU Hao, LI Tianming, LIU Jinlei, et al. Construction of a marker-free deletion system based on the uracil phosphoribosyl transferase gene as a negative selection marker in Ketogulonigenium vulgare[J]. Food Science, 2017, 38(4): 39-44. (in Chinese with English abstract)
DOI:10.7506/spkx1002-6630-201704007. http://www.spkx.net.cn
收稿日期:2016-04-07
基金项目:国家高技术研究发展计划(863计划)项目(2014AA022102)
作者简介:陆浩(1991—),男,硕士研究生,研究方向为合成生物学与代谢工程。E-mail:840835694@qq.com
*通信作者:冯惠勇(1967—),女,教授,硕士,研究方向为合成生物学与代谢工程。E-mail:fenghuiyong@hotmail.com