
图1 样品多样性指数分析
Fig. 1 Analysis of microbial diversity index
臧凯丽1,江 岩1,孙 勇2,*,闫亚丽1,*,陈庆森1,赵林森3,赵 培1,崔文静3,马新颖3
(1.天津商业大学生物技术与食品科学学院,天津市食品生物技术重点实验室,天津 300134;2.北京食品科学研究院,中国肉类食品综合研究中心,北京 100068;3.河北一然生物科技有限公司,河北 石家庄 050800)
摘 要:为探究益生菌剂对肠道疾病人群菌群多样性及丰度水平的调节能力。本研究以健康、便秘、腹泻及腹胀、肠易激综合征患者、排便不规律等人群为对象,使其定时、定量摄入益生菌剂(probotic preparations,PPrs)共6 周,采集新鲜粪便样品并提取DNA,利用Ion Torrent PGM测序技术进行16S rRNA V3区扩增子测序,并联合生物信息学和多变量统计学分析方法对测序数据进行多样性分析。在测序深度满足要求的情况下,从门、科、属乃至种4 个水平由浅至深挖掘益生菌剂对各受试人群肠道菌群失调的调节作用。所有测序序列在97%相似水平划分得到2 320 个分类操作单元,拟杆菌门(Bacteroidetes)、硬壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)为各组的优势菌门,占总序列数的99.81%。益生菌剂对各组受试人群的共性特征是:毛螺菌科(Lachnospiraceae)的调整均有长期的积极作用,而产碱杆菌科(Alcaligenaceae)、理研菌科(Rikenellaceae)、双歧杆菌科(Bifidobacteriaceae)等优势菌科在干预停止后又恢复至干预前的状态。益生菌剂对各种疾病状态人群的作用存在差异,便秘组的蓝细菌门(Cyanobacteria)和梭杆菌门(Fusobacteria)均从无到有。经主成分分析(principal component analysis,PCA)鉴定显示,在干预第1~4周期间,益生菌剂对腹泻组属水平的调整优于其他两组,柔嫩梭菌属(Faecalibacterium)、不动杆菌属(Acinetobacter)、布劳特氏菌属(Blautia)、梭菌属(Clostridium)、小类杆菌属(Dialister)、埃格特菌属(Eggerthella)、颗粒链菌属(Granulicatella)、乳杆菌属(Lactobacillus)、草酸杆菌属(Oxalobacter)、Pyramidobacter等10 个属多样性及相对丰度均有明显的提升;便秘组仅有普氏菌属(Prevotella)、巨单胞菌属(Megamonas)、柯林斯菌属(Collinsella)3 个属相对丰度变化;其他组的Adlercreutzia、Collinsella、克雷白氏杆菌属(Klebsiella)、Parabacteroides、萨特氏菌属(Sutterella)5 个属有相对丰度的变化。PCA分析还揭示了益生菌剂在不同肠道疾病个体菌群结构的调整方面存在明显的差异,对不同肠道疾病的改善及治疗作用也不一样。另外,便秘组的Bacteroides和Odoribacter有降低的趋势,且回复到与对照组接近的状态。经热图分析鉴定发现,Akkermansia muciniphila、Bacteroides fragilis、Faecalibacterium prausnitzii等几种已被认定功效的关键菌阶段性地出现,但是停止干预后即消失,原有的Bacteroides ovatus开始降低,这些菌的出现对维护宿主健康具有价值。综上所述,该益生菌剂干预能起到改变肠道微生物多样性的功效,并具有调整菌群丰度水平的作用,表现出了抑制肠道有害微生物生长的作用,特别是对肠道健康有益菌的出现更体现出该益生菌剂的生理功效;因此,该益生菌剂应用于维持肠道菌群结构的稳态具有广阔的市场前景。
关键词:益生菌剂;肠道疾病;菌群丰度;Ion Torrent PGM测序技术;生物信息学
肠道既是人体的消化场所又是人体最大的免疫器官,富集着庞大而复杂的微生物群落,约有800余种7 000多株的细菌,在亚种或菌株水平上表现出极大的多样性[1]。肠道微生物对人类健康有着重要的影响,能够帮助宿主代谢,为肠道上皮细胞提供营养,增强免疫功能,抵抗外来微生物的入侵[2]。正常状态下,肠道微生物与宿主间存在着复杂而微妙的动态平衡,微生物从宿主汲取生长所需的养分,宿主则借助微生物来降解一些自身不能分解利用的物质[3]。肠道微生物与宿主之间的平衡一旦被打破,就会造成肠道菌群结构紊乱,肠道微生物就会从能量吸收、内毒素血症、短链脂肪酸、胆碱、胆汁酸代谢和脑肠轴等多种途径影响宿主的健康[4]。肠道菌群之所以被称为人类的“第二指纹”,是由于每个个体都有自己独特的肠道微生物组,其组成和功能通常会随着宿主的基因、饮食及环境等因素的变化而改变[5]。早在20世纪90年代初,Metchnikoff就提出了著名的“梅氏假说”,发现保加利亚人长寿的秘诀在于其长期饮用发酵乳[6]。近些年来,由于益生菌能够直接或者间接地调整宿主肠道微生物的组成、增加有益菌的比例,激活宿主内源性微生物或者免疫系统的活性,被广泛用于预防和治疗许多肠道疾病,尤其针对腹泻、便秘及肠易激综合征等病症。目前研究较多的益生菌主要包括乳酸杆菌、乳酸链球菌、粪链球菌、芽孢杆菌、双歧杆菌和酵母菌等。Chen等[7]研究发现,酪酸梭菌、粪链球菌、芽孢杆菌三联剂Bio-Three具有提高治疗腹泻疗效,缩短病程等效果;Heo等[8]研究发现,地衣芽孢杆菌(Bacillus licheniformis)能够通过抑制相关病原菌生长,安全有效地治疗各种腹泻和肠道内异常发酵。Guandalini等[9]通过对受试人群每天服用混合8 种冻干菌剂的研究发现,混合冻干菌剂显著地改善了肠易激综合征的病症;丁圣等[10]通过对小鼠模型的研究发现,乳酸杆菌和低聚果糖对肠道内的肠杆菌、肠球菌等有害菌有抑制作用,对双歧杆菌等肠道有益菌具有增殖作用。虽然益生菌剂的相关研究颇多,但大多以其作用机制为出发点研究其对肠道微生物多样性以及菌群结构的调节作用,关于具体菌群丰度的调节作用的报道较少。
随着该领域研究的深入,宏基因组学测序技术已经从传统的Sanger双脱氧链终止法测序技术发展到第二代高通量测序,目前基于16S rRNA基因可变区高通量测序的技术应用较多的有Roche454、Illumina的HiSeq及MiSeq、Life的Ion Torrent高通量测序技术[11],这些技术为解析肠道菌群丰度的变化与人类健康的关系做出了贡献。
鉴于前人对益生元、益生菌及合生元等益生特性的研究[12],本研究采用对便秘、腹泻等肠道疾病有明显调节作用的菊粉、水苏糖、植物乳杆菌、嗜酸乳杆菌、干酪乳杆菌、两歧双歧杆菌等成分复合制备而成的益生菌剂对不同健康状态的受试人群加以干预,从调整菌群结构丰度水平、提高菌群多样性和改善肠道微生物与宿主平衡的角度出发,分析益生菌剂对肠道相关疾病的作用,为以肠道微生物为靶点的肠道疾病的预防提供理论依据,并提供新的思路。
益生菌剂:抗性糊精(15%~20%,质量分数,下同)、菊粉(10%~15%)、水苏糖(5%~10%)、麦芽糊精(10%~15%)、植物乳杆菌LP45(Lactobacillus plantarum)、嗜酸乳杆菌La28(Lactobacillus acidophilus)、干酪乳杆菌YMC1069(Lactobacillus casei)、两歧双歧杆菌TMC3115(Bifidobacterium bifidum),活菌数比例为1∶1∶1∶1,活菌数2×1010CFU/g,由河北一然生物科技有限公司提供。
Ion Xpress™ Barcode Adapters Kit、Ion PGM™Template OT2 200 Kit、Ion PGM™ Sequencing 200 Kit v2、Ion Plus Fragment Library Kit、Ion Library TaqMan™Quantitation Kit、Pfu DNA聚合酶 美国Thermo Scientific公司;Qubit®dsDNA HS Assay Kit 美国Invitrogen公司;High Sensitivity DNA Kit 美国Agilent公司;QIAamp Fast DNA Stool Mini Kit 德国QIAGEN公司;MiniBEST Agarose Gel DNA Extraction Kit 宝生物工程(大连)有限公司。
3K18高速冷冻离心机 美国Sigma公司;DYY-2C电泳仪 北京六一仪器厂;508-U001 Ion Torrent测序平台、2.0 Qubit荧光定量仪 美国Invitrogen公司;Ion One TouchTM2INS(模板制备系统)、Ion One Touch™ES(模板富集系统)、磁力架 美国Thermo Scientific公司;2100生化分析仪 美国Agilent公司;GeneAmp 7900聚合酶链式反应仪 美国ABI公司;II-3 Biowave DNA紫外-可见分光光度计 英国柏诺公司。
1.3.1 受试人群健康特征描述
受试人群年龄为18~35 岁。健康、便秘、腹泻志愿者的临床症状确定:通过对受试者的询问及其自身表述,按照临床医学给定的便秘、腹泻以及健康个体的临床症状确定;要求志愿者在开始实验的两个星期内未发生任何疾病;实验期间无使用抗生素等药物或接受治疗的情况,同时不限制受试人群每日饮食内容及时间;另外,还要求受试者3 个月内未参与其他实验研究。
1.3.2 受试人群的分组及样品采集
本实验从38 名志愿受试人群中[13],深度挖掘益生菌剂对不同疾病个体肠道菌群多样性以及丰度水平的变化,故在已有样本的条件下,选择男性5 名,女性12 名。受试人群保持饮食及睡眠正常且无限制,每日服用益生菌剂,早晚各一次,2 g/次(活菌数约2×1010CFU/g);干预期间记录饮酒、服药等特殊情况。服用益生菌剂前统一采集粪便1 次,干预期间每周采集2 次,共6 周,停止服用2 周后,采集2 次,并将其统一放置于液氮罐中保存,共采集粪便样品180 个。用QIAamp Fast DNA Stool Mini Kit提取粪便细菌基因组DNA,用1%琼脂糖凝胶电泳检测后,又根据细菌基因组完整性以及浓度等判断,约有104 个样本符合要求,保存备用。最后选择健康(1男2女)、便秘(2女)、腹泻(1男2女)、排便不规(1男)、肠易激综合征(1男)、腹胀(1女)等不同生理状态11 人共69 个样品进行高通量测序,按健康状况将样品依次分为健康对照组(CT)、便秘组(CO)、腹泻组(DI)及其他组(OT)共4 组。
1.3.3 肠道菌群16S rRNA V3区的高通量测序
首先,进行粪便中微生物总DNA的提取,然后对肠道菌群16S rRNA V3区进行聚合酶链式反应扩增,通过Ion Torrent PGM进行测序[13]。
1.3.4 测序数据生物信息学和多变量统计学分析
首先,用FastQC将原始数据进行质控,并利用NGStool kits过滤掉低质量序列。将质控得到的高质量序列根据样品Barcode和引物进行数据的正、反方向分选,进入微生物生态学定量研究(quantitative insights into microbial ecology,QIIME)后再次进行质控以及去除嵌合体,利用usearch方法在97%的相似水平下划分分类操作单元(operational taxonomic units,OTU);选取每个OTU中丰度最高的序列为代表序列,再参照样品的信息,生成以序列数代表的每个样本中每个OTU丰度的OTU Table。然后,以OTU Table为基础按照进行物种分类,生成系统发育树,并对样品各水平相对丰度进行划分;依据OTU Table数据进行每个文库多样性分析,并利用R软件(Version 3.4.2)绘制稀释曲线、Rank-Abundance曲线、Chao1指数箱线图以及Shannon指数箱线图。最后,为了减少因某些样本中含有丰度极高的OTU而造成的样本间距离分布异常和测序时带来的偏差,研究中对所有样本中的OTU丰度进行转化,利用转换后的数据即样品中各进化水平相对丰度百分比用Stamp软件寻找差异OTU,并用R软件(Version 3.4.2)绘制后续的热图、堆砌条图以及实现降维的主成分分析(principal component analysis,PCA)。
2.1.1 多样性指数分析
图1 样品多样性指数分析
Fig. 1 Analysis of microbial diversity index
由图1a可知,随着样品测序深度的增加,观测物种指数曲线趋于平缓,说明当前测序深度足以发现各样本生境中的大部分物种。此外,从图1b中反映物种丰富度及均匀度的Rank-Abundance曲线可以看出,随着OTU等级的增加,曲线愈加平坦并且在横轴的跨度也越来越大,说明样品中物种的均匀度和相对丰度都达到了很高的水平。
2.1.2 益生菌剂干预下的肠道菌群多样性的变化

图2 益生菌剂干预下肠道菌群α多样性的变化
Fig. 2 Changes in α diversity of intestinal flora with the intervention of probiotics
图2 a、b分别反映受试人群肠道菌群物种丰富度和微生物多样性,与CT组相比,3 组不同健康状态的受试人群肠道菌群的物种丰富度在益生菌剂干预后都有不同程度的增加,并且都向CT组靠近,说明益生菌剂干预对不同健康状态人群肠道菌群的多样性具有一定影响。
以97%的相似水平通过usearch划分OTU进化水平,在门水平上选取样品中最大丰度排名前十的物种划分为10 个菌门,结果如图3所示。
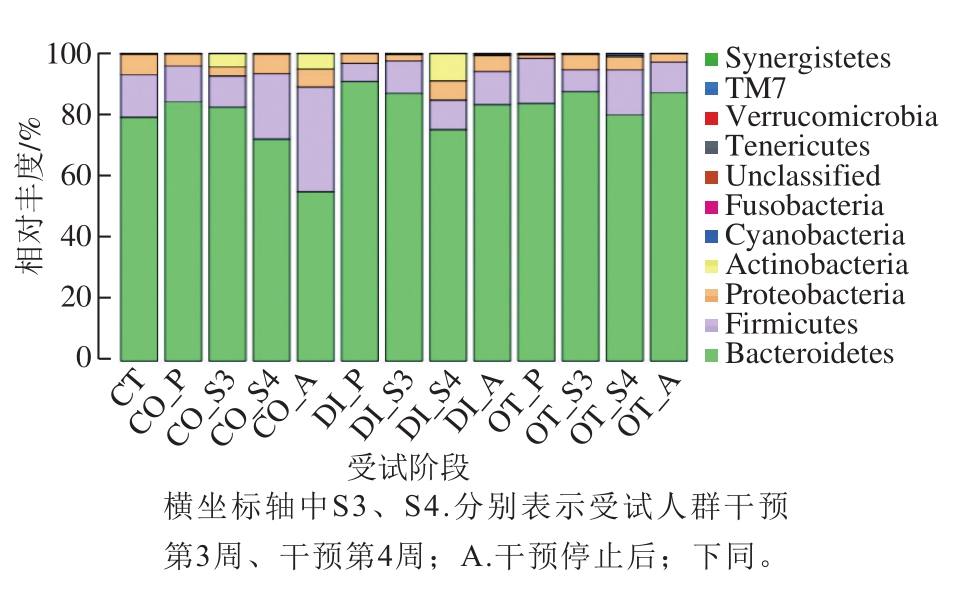
图3 益生菌制剂对肠道菌群门水平上相对丰度的影响
Fig. 3 Effect of probiotics on relative abundance of dominant phyla
图3显示所有OTU被划分为10 个门,其中拟杆菌门(Bacteroidetes)、硬壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)为各组受试人群肠道菌群的优势菌门,占总序列数的99.81%,其中Bacteroidetes的含量最丰富,约占总序列数的80.30%,其次是Firmicutes,约占总序列数的13.14%,而Proteobacteria和Actinobacteria的占比分别为4.14%、1.98%。其余的如蓝细菌门(Cyanobacteria)、梭杆菌门(Fusobacteria)、互养菌门(Synergistetes)、TM7、疣微菌门(Verrucomicrobia)及无壁菌门(Tenericutes)在各样品中所占比例不足0.1%,另外,还有约0.01%左右的细菌(Unclassified)无法确定具体分类地位。
从图3可以看出,不同健康状况的受试人群肠道菌群结构中门水平相对丰度的差异较明显,从便秘组看,受试人群肠道菌群结构中Bacteroidetes、Firmicutes、Proteobacteria这3 种优势菌门有一个共同特点:在干预4 周后的菌群相对丰度分别为80.07%、12.48%、7.27%,与健康对照组(79.32%、13.75%、6.63%)最为接近,不同的是,Bacteroidetes的相对丰度随着益生菌剂的干预逐渐降低,由最初的84.27%降到44.78%;Firmicutes相对丰度在益生菌剂干预阶段虽略有波动,但是总体呈上升趋势;Proteobacteria相对丰度随益生菌剂干预时间的延长呈先降低后升高起伏波动状态。此外,Cyanobacteria和Fusobacteria的相对丰度与健康对照组(0.08%、0.005%)具有一定差异,但是,在益生菌剂干预第4周均从零上升到0.001 7%,不过在益生菌剂停止干预后又恢复到了干预前的状态。对于腹泻组受试人群来说,益生菌剂对Bacteroidetes、Firmicutes和Proteobacteria有积极的调整作用,除Proteobacteria在干预4 周后由最初的3.08%调整到6.87%,与对照组(6.63%)最为接近外,其余两种均在干预停止后保持与对照组接近的84.17%、8.06%状态,同时,Fusobacteria在停止干预后异常升高至0.39%,远超对照组(0.005%)。就其他健康状况的受试人群来说,Bacteroidetes和Firmicutes在干预第4周分别调整到80.05%、14.65%,Proteobacteria和Actinobacteria在干预3周前分别由1.12%、0.03%升至4.94%、0.29%,与对照组(6.63%、0.21%)最为接近,但是,在停止干预后,Firmicutes、Proteobacteria及Actinobacteria并没有保持不变而是呈现降低的趋势。
此外,对排名前20的各个菌科在益生菌剂干预下的各个样品中相对丰度进行比较发现,Firmicutes的毛螺菌科(Lachnospiraceae)、瘤胃球菌科(Ruminococcaceae)、韦荣球菌科(Veillonellaceae)、梭菌科(Clostridiaceae),Bacteroidetes的普雷沃氏菌科(Prevotellaceae)、理研菌科(Rikenellaceae)、拟杆菌科(Bacteroidaceae)、紫单胞菌科(Porphyromonadaceae),Proteobacteria的肠杆菌科(Enterobacteriaceae)、产碱杆菌科(Alcaligenaceae)、弧菌科(Desulfovibrionaceae),以及Actinobacteria的双歧杆菌科(Bifidobacteriaceae)在科的水平上相对丰度占优势地位。
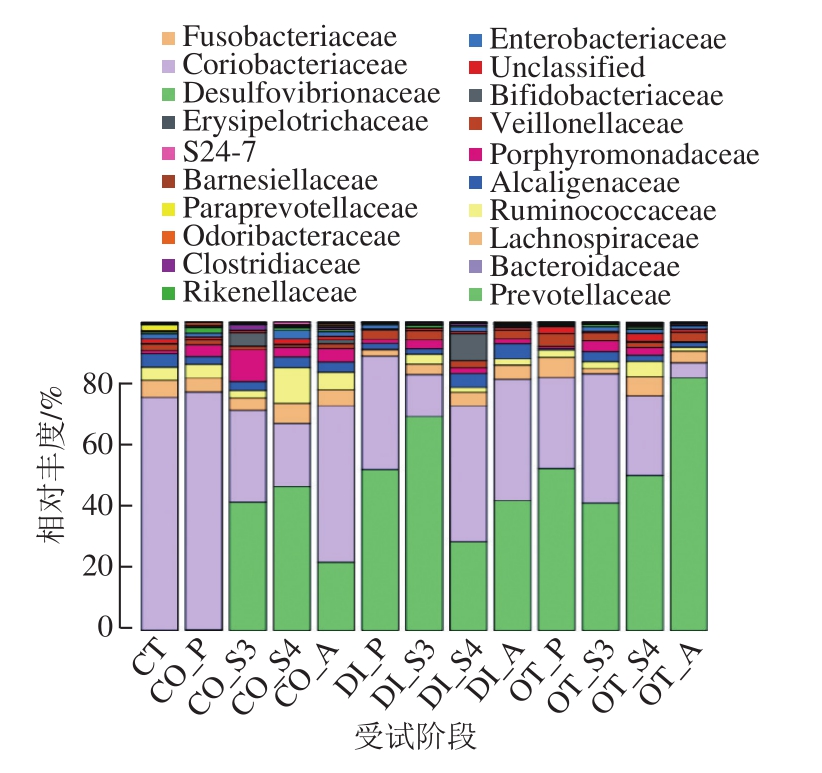
图4 益生菌制剂对肠道菌群科水平上相对丰度的影响
Fig. 4 Effect of probiotics on relative abundance of dominant families
从图4中可以看出,受试人群在益生菌剂的干预下,便秘组的Prevotellaceae、Lachnospiraceae、Alcaligenaceae、红蝽菌科(Coriobacteriaceae)及S24-7相对丰度在干预第4周后分别由0.23%、4.43%、2.58%、0.005%、0.03%增加至46.47%、6.50%、3.45%、0.1%、0.8%,停止干预后又降至22.01%、5.09%、3.28%、0.06%、0.29%;Bacteroidaceae、Desulfovibrionaceae和Rikenellaceae相对丰度分别由76.97%、0.085%、1.85%降至20.65%、0.023%、0.72%,停止干预后又升至50.75%、0.11%、0.83%。其中,Alcaligenaceae、Rikenellaceae、Coriobacteriaceae在向对照组(4.22%、0.58%、0.088%)靠近。腹泻组中,受试人群中Lachnospiraceae、Alcaligenaceae、Bifidobacteriaceae、Clostridiaceae、Coriobacteriaceae、Erysipelotrichaceae的菌群相对丰度从开始干预到第4周分别由1.87%、1.73%、0.12%、0.15%、0.05%、0.07%增至4.39%、4.46%、8.65%、0.55%、0.18%、0.23%;而Veillonellaceae经益生菌剂干预后由3.06%降至2.16%。其中,Lachnospiraceae、Alcaligenaceae、Erysipelotrichaceae、Veillonellaceae向对照组(5.49%、4.22%、0.24%、2.20%)状态调整,并且,Lachnospiraceae在干预停止后不会恢复至干预前的状态,仍保持在4.70%左右。就其他组受试患者菌群结构而言,在益生菌剂的作用下,除Veillonellaceae有下降的趋势外,Erysipelotrichaceae、Bifidobacteriaceae、Rikenellaceae、Odoribacteraceae、Desulfovibrionaceae、Coriobacteriaceae、S24-7都呈现增长的趋势,其中,Enterobacteriaceae、Bifidobacteriaceae、Rikenellaceae、Odoribacteraceae、Desulfovibrionaceae分别从干预前的0.19%、0.003 3%、0.32%、0.23%、0.17%提升至1.50%、0.19%、0.59%、0.38%、0.20%,接近于对照组相对丰度(1.49%、0.12%、0.58%、0.44%、0.28%)。
根据上述肠道微生物整体结构的分析发现,各疾病状态受试人群的肠道微生物多样性与对照组具有一定程度的差异,益生菌剂的干预使得其菌群结构的组成发生了改变,为了寻找益生菌剂干预前后造成肠道微生物多样性差异的关键菌属,进一步在属水平上进行Welch’s t-test检验显著性差异,进而进行Stamp分析,发现3 组受试人群的肠道微生物经益生菌剂干预后在属水平均有显著不同,结果如图5所示。
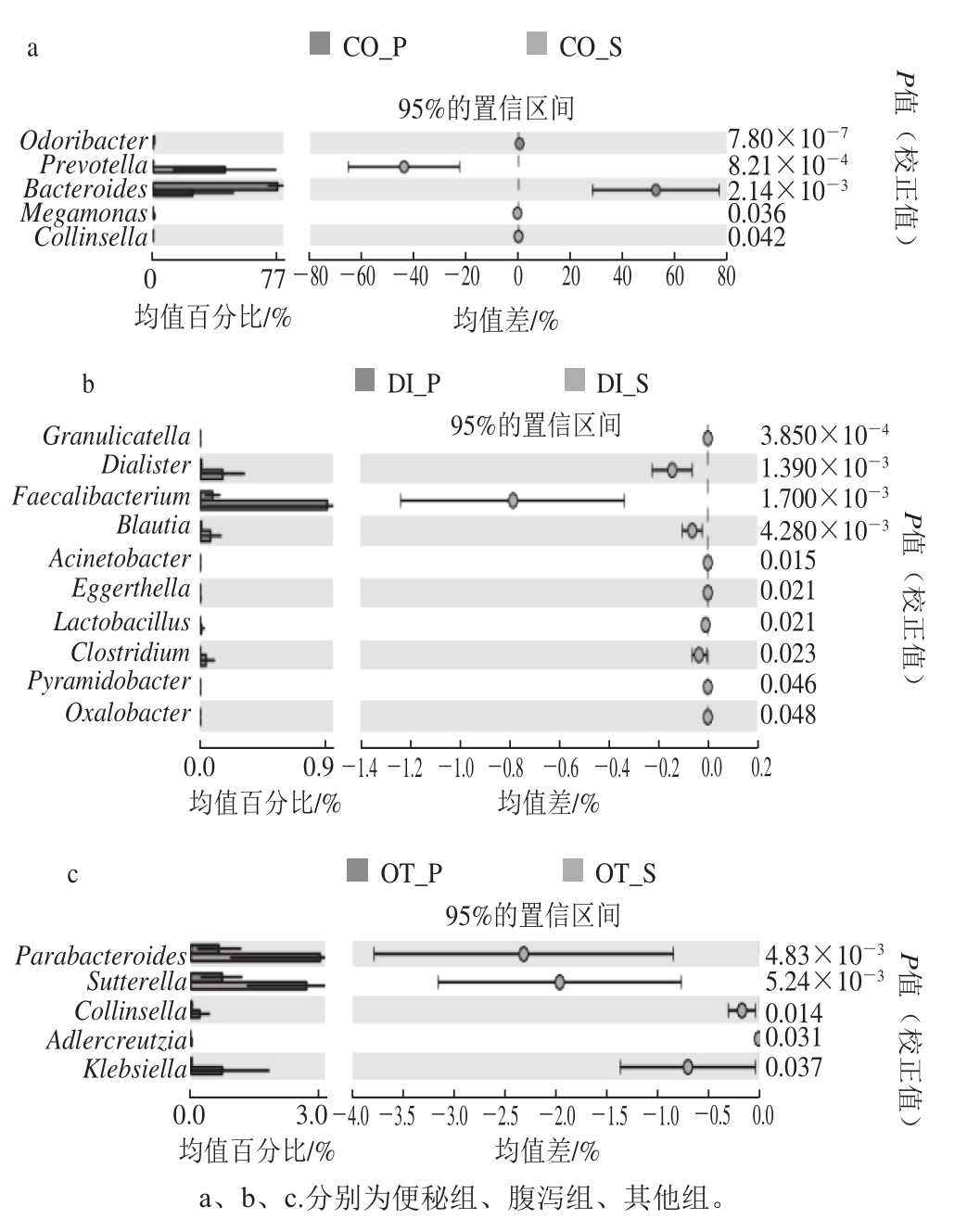
图5 肠道菌群中关键菌属的变化
Fig. 5 Change of key bacterial genus in intestinal flora
从图5中可以看出,3 组受试人群在益生菌剂干预下共有20 个关键菌属存在显著差异(P<0.05),其中,便秘组有5 个,包括拟杆菌属(Bacteroides)、柯林斯菌属(Collinsella)、巨单胞菌属(Megamonas)、Odoribacter、普氏菌属(Prevotella);腹泻组有柔嫩梭菌属(Faecalibacterium)、不动杆菌属(Acinetobacter)、布劳特氏菌属(Blautia)、梭菌属(Clostridium)、小类杆菌属(Dialister)、埃格特菌属(Eggerthella)、颗粒链菌属(Granulicatella)、乳杆菌属(Lactobacillus)、草酸杆菌属(Oxalobacter)、Pyramidobacter等10 个,其他组有5 个,即Adlercreutzia、Collinsella、克雷白氏杆菌属(Klebsiella)、Parabacteroides和萨特氏菌属(Sutterella)。
为了探究关键菌属对肠道菌群整体结构的影响,通过Stamp分析寻找到20 个关键菌属,基于20 个关键菌属进行PCA分析,结果如图6所示。
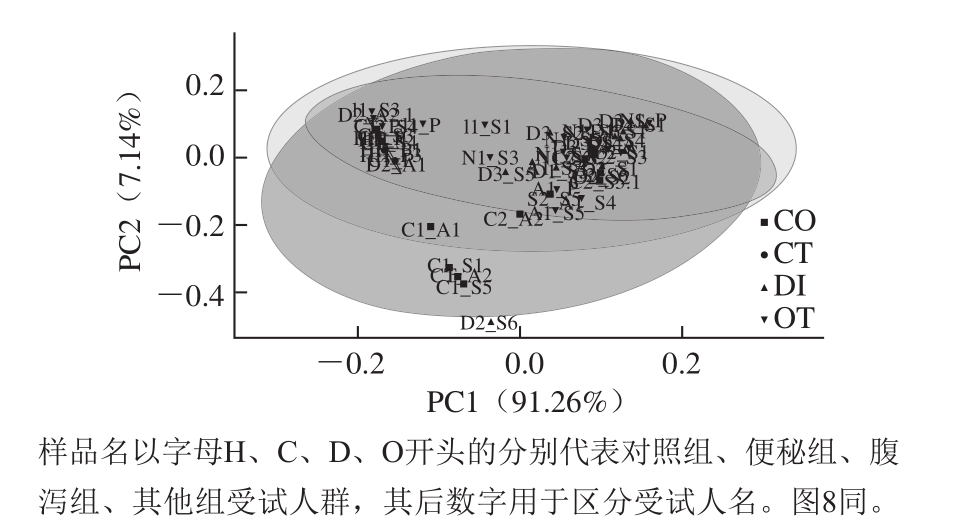
图6 PCA分析
Fig. 6 PCA analysis
图6 显示,3 组不同健康状态样品均与健康对照组呈现交叠的状态,各组样品并不能完全分开,说明相较其他因素,益生菌剂对肠道菌群结构的调节作用较为明显。并且,3 组样品在X轴上的距离比较长。随着益生菌剂干预时间的延长,便秘组关键菌属由最初接近健康对照组逐渐沿着PC1(贡献率91.26%)、PC2(贡献率7.14%)拉开距离,在PC2方向尤为明显,但是距离健康对照组越来越远;腹泻组菌属则在益生菌剂干预第5周直至停止干预后沿PC1逐渐向对照组接近;其他组菌属在益生菌剂干预期间向健康对照组靠近,在停止干预后并没有继续保持这种状态。在PCA图中,各组样品随着益生菌剂干预的时长呈现调整状态,正是这种差异显著的状态,从另一层面上说明这20 个菌属可能是造成疾病组与健康对照组受试人群肠道菌群结构差异的关键菌属。
将Welch’s t-test检验出的存在显著差异的20 种菌进行了相对丰度柱状图的绘制,如图7显示,在益生菌剂的干预下,便秘组Bacteroides、Prevotella、Parabacteroides的变化明显且迅速,在干预第1周开始分别由最初的76.98%、0.23%、3.69%调整至22.18%、35.13%、18.7%,Megamonas由最初的0.005%逐渐向健康对照组(0.91%)调整,在第3周时达到1.06%,与对照组最为接近,Collinsella也在干预第3周开始出现;腹泻组Lactobacillus和Clostridium从无到有,并且,Clostridium在第1周就达到了0.06%,与健康对照组的相对丰度相同,Lactobacillus在第4周时达到0.006 7%,与健康对照组(0.007 5%)最为接近;此外,益生菌剂具有升高Blautia降低Dialister和Faecalibacterium的作用,其中,Blautia由最初的0.003 3%升至0.05%,并且,干预停止后并没有恢复至干预前的状态;就其他组而言,益生菌剂对5 种关键菌属具有升高的作用,Adlercreutzia在干预第1周从无到有,升高了0.005%,Collinsella、Klebsiella、Parabacteroides和Sutterella从干预开始至停止干预一直处于升高的趋势。

图7 益生菌制剂对肠道菌群属水平上相对丰度的影响
Fig. 7 Effect of probiotics on relative abundance of dominant genus

图8 物种相对丰度聚类热图
Fig. 8 Species abundance from clustering heatmap
为探究益生菌剂对肠道微生物种水平的影响,对其相对丰度进行聚类,得相对丰度聚类热图,图8展示了物种间的相似性和相对丰度以及样本间的相似性。
从图8可以看出,受试样品多以Bacteroides为核心菌属,说明肠型之说的存在。经益生菌剂干预,便秘组受试人群肠道中,玉米乳杆菌(Lactobacillus zeae)在干预第3周相对丰度达到最高(0.02%),超过健康对照组(0.01%)。Akkermansia muciniphila和两形真杆菌(Eubacterium biforme)在干预停止后的第2周相对丰度最高(0.42%、0.02%),脆弱拟杆菌(Bacteroides fragilis)、柔嫩梭菌(Faecalibacterium prausnitzii)的相对丰度在第7周(16.17%、1.95%)及干预停止的第2周(11.6%、2.99%)较高,活泼瘤胃球菌(Ruminococcus gnavus)在干预第4周及干预停止的第2周相对丰度升高,由最初的0.02%增至0.04%、0.08%;此外,卵形拟杆菌(Bacteroides ovatus)和Prevotella stercorea在干预前相对丰度最高,这说明益生菌剂具有降低二者相对丰度的作用。在腹泻组受试人群肠道菌群中,长双歧杆菌(Bifidobacterium longum)和粪便拟杆菌(Bacteroides stercoris)在干预第6周(9.09%、0.33%)相对丰度最高,Actinobacillus parahaemolyticus和副流感嗜血杆菌(Haemophilus parainfluenzae)在干预第1周(0.12%、0.07%)时相对丰度增加明显,Anoxybacillus kestanbolensis的相对丰度由最初不存在到第4周(0.07%)最高,短乳杆菌(Lactobacillus brevis)、Blautia producta及细长真杆菌(Eubacterium dolichum)都由最初不存在到停止干预后相对丰度达到最高(0.01%、0.03%、0.09%)。对于其他组,腹胀受试患者(A1_S6)的瘤胃乳酸杆菌(Lactobacillus ruminis)在益生菌剂干预第6周时相对丰度(0.12%)最高;肠易激综合征患者(I1_S1、I1_S2、I1_S4)及排便不规律患者(N1_S6)的Bacteroides coprophilus在第1~6周时开始出现。
由于肠道微生物可以吸收宿主自身难以消化吸收的成分及分泌到肠腔中的物质,所以肠道微生物的组成会随机体摄入的食物成分的改变而变化。也就是说,膳食结构是影响肠道菌群结构和功能最为重要的因素,不当的饮食会扰乱肠道微生物和宿主之间的稳态平衡,造成肠道菌群失调进而诱发多种肠道疾病,益生菌剂作为一种常见食品被用于调节肠道菌群结构可有效地预防甚至是治疗相关疾病[14]。
本实验通过对受试人群粪便样本的测序和解析发现,益生菌剂对便秘、腹泻和其他症状受试人群肠道菌群多样性及物种相对丰度均有积极的调整作用,都使之向健康状态靠近。在门水平上,各组受试人群肠道菌群以Bacteroidetes、Firmicutes、Proteobacteria、Actinobacteria为优势菌门,与前人研究中这4 种优势菌门达90%以上的结论[15]一致,造成肠道微生物门水平多样性较低的原因有两个,一方面源于宿主对胃肠道微生物组成及其功能的选择性压力,另一方面可能是与地球漫长的历史相比,人类进化史相对较短而造成的[16]。但是,肠道微生物的相对丰度在益生菌剂干预期间乃至干预停止后起到了调整作用,符合Murphy等[17]得出的“改变饮食可以影响肠道微生物的结构”的结论。其中,便秘组受试人群中Bacteroidetes、Firmicutes、Proteobacteria这3 种优势菌门在益生菌剂干预第4周时达到与对照组最接近的状态,这说明益生菌剂能够在较短的时间影响肠道菌群的结构。并且,在整个干预阶段,Bacteroidetes的相对丰度随着益生菌剂的干预由最初的84.27%降到44.78%;Firmicutes虽略有波动,但是总体呈上升趋势;Proteobacteria相对丰度随益生菌剂干预时间的延长呈先降低后升高的起伏波动状态。这提示益生菌剂能够降低便秘人群中起促炎作用的Bacteroidetes,升高Firmicutes的相对丰度。Cyanobacteria和Fusobacteria在益生菌剂的干预下达到了从零到0.001 7%的状态;但是,益生菌剂停止干预后又恢复到了干预前的状态,这也有力地支持了服用益生菌制品对肠道菌群结构以及对宿主健康状况的作用存在短期功效的事实。同样,益生菌剂对腹泻组受试人群的Bacteroidetes、Firmicutes和Proteobacteria表现积极的调整作用,除Proteobacteria在干预4 周后与健康对照组(6.87%)最为接近外,其余两种均在干预停止后保持在84.17%、8.06%,与健康对照组接近。就其他健康状况的受试人群来说,Bacteroidetes和Firmicutes在干预第4周以及Proteobacteria和Actinobacteria在干预3周前与对照组最为接近,说明益生菌剂具有调整其他组健康状况人群的Bacteroidetes和Firmicutes、提高Proteobacteria和Actinobacteria相对丰度的作用。但是,在停止干预后,Firmicutes、Proteobacteria及Actinobacteria并没有保持不变而是呈现降低的状态。以上都体现了益生菌剂一方面补充肠道的正常菌群或促进有益菌的增殖,另一方面拮抗病原菌[18]。在短期内肠道菌群相对丰度相应改变也说明肠道微生物能够对益生菌剂的介入迅速做出反应,这可能与各菌门的代谢产物(如短链脂肪酸)有关,肠道微生物在肠道内将多糖降解成单糖和短链脂肪酸,提高能量利用效率,而短链脂肪酸是结肠和小肠上皮细胞的主要供能物质,能够较强地维护肠道形态及功能,并对肠道疾病具有一定的治疗作用,也是研究益生菌剂对肠道微生物影响的重要指标[19]。有报道指出,Bacteroidetes以生成乙酸和丙酸为主,而Firmicutes以生成丁酸为主[20]。Bacteroidetes首先将丙酮酸氧化成乙酰辅酶A,然后经磷酸乙酰转移酶、乙酸激酶生成乙酸[21];此外,Bacteroidetes生成丙酮酸还可通过丙烯酸途径形成丙酸[22]。Firmicutes首先将丙酮酸转化成乙酰辅酶A,然后通过丁酰辅酶A的生成等一系列途径生成丁酸[23]。乙酸和丙酸可抑制肿瘤坏死因子-α的释放,对结肠炎有良好的治疗效果;丁酸能有效抑制结肠肿瘤细胞增殖、诱导肿瘤细胞分化的凋亡、影响原癌基因的表达,从而预防和治疗结肠癌[24-26]。但是,停止干预后部分肠道微生物向干预前状态恢复的现象与Carmody等[27]提出的“肠道菌群存在一种记忆效应”之说有关,即在饮食(如益生菌剂)干预后,它的组成和代谢状况除了依赖于当前的饮食,也少部分依赖于它之前的饮食,正如Zhang Chenhong等[28]的研究结果,这并不意味着无论如何破坏菌群结构都能使之得到恢复。
其次,就科水平而言,3 组受试人群中这些优势菌科的相对丰度均有调整,但是,在干预第4周时部分菌科相对丰度调整较好。便秘组Alcaligenaceae、Rikenellaceae、Coriobacteriaceae由最初的2.58%、1.85%、0.005%分别调整至3.45%、0.72%、0.1%,接近于健康对照组(4.22%、0.58%、0.088%);腹泻组Lachnospiraceae、Alcaligenaceae、Erysipelotrichaceae及Veillonellaceae由最初的1.87%、1.73%、0.07%、3.06%分别调整至4.39%、4.46%、0.23%、2.16%,接近于健康对照组(5.49%、4.22%、0.24%、2.20%);并且,Lachnospiraceae在停止干预后仍保持在4.70%左右;其他组Enterobacteriaceae、Bifidobacteriaceae、Rikenellaceae、Odoribacteraceae、Desulfovibrionaceae分别从干预前的0.19%、0.0033%、0.32%、0.23%、0.17%提升至1.50%、0.19%、0.59%、0.38%、0.20%,接近于健康对照组相对丰度(1.49%、0.12%、0.58%、0.44%、0.28%)。以上结果提示益生菌剂能够通过提高便秘组受试人群的Alcaligenaceae和Coriobacteriaceae并降低Rikenellaceae,提高腹泻组的Lachnospiraceae、Alcaligenaceae和Erysipelotrichaceae并降低Veillonellaceae,提高其他组Enterobacteriaceae、Bifidobacteriaceae、Rikenellaceae、Odoribacteraceae、Desulfovibrionaceae来改善病理状态人群肠道菌群相对丰度,这可能与菌科本身所产生的代谢产物功能相关,Enterobacteriaceae、Lachnospiraceae、Veillonellaceae、Clostridiaceae以及Bifidobacteriaceae这几个菌科与短链脂肪酸的产生相关,尤其是Lachnospiraceae中的Blautia、Coprococcus、Lachnospira以及Roseburia都能参与到将碳水化合物分解为短链脂肪酸的代谢过程中,特别是Roseburia、Blautia以及Lachnospira主要参与丁酸盐的产生,丁酸盐不仅是上皮细胞的能量来源,可以通过组蛋白的高度乙酰化和抑制核因子-κB信号通路来作为肠黏膜促炎细胞因子表达的抑制剂,还可以促进黏蛋白和抗菌肽的产生来增强肠屏障功能,通过直接提高紧密结合蛋白的表达来增强肠上皮细胞的完整性,影响细胞的功能和结肠的健康,具有抗癌和抗炎的潜在功能,这些菌的减少往往会引起丁酸盐的减少,Enterobacteriaceae可能与炎症和胰岛素抵抗有关,梭菌(Clostridium)可利用多种氨基酸,相应生成多种有机酸,通过丙烯酸途径由丙氨酸产生丁酸,也可在苏氨酸脱水酶和酮酸脱氢酶的参与下由苏氨酸产生丙酸[29-30]。此外,益生菌剂停止干预后各菌群相对丰度又逐渐向干预前恢复,这再次说明益生菌剂具有短期调整肠道菌群的效力。
然后,对属水平进行Welch’s t-test检验及Stamp分析发现有20 个存在显著差异的菌属,经PCA鉴定,它们也是造成不同健康状况组与健康对照组间存在显著差异的关键菌属,提示益生菌剂干预能够引起20 个关键菌属发生显著变化。便秘组样品随着益生菌剂干预时间的延长距离健康对照组越来越远;腹泻组在益生菌剂干预时直至停止干预后都逐渐向健康对照组接近;其他组样品在益生菌剂干预期间也表现向健康对照组靠近的趋势,但是在停止干预后并没有继续保持这种状态,这提示益生菌剂对腹泻组的调整效果优于其他两组。此外,研究结果显示,在益生菌剂的干预下,便秘组Prevotella、Megamonas、Collinsella,腹泻组Faecalibacterium、Acinetobacter、Blautia、Clostridium、Dialister、Eggerthella、Granulicatella、Lactobacillus、Oxalobacter、Pyramidobacter以及其他组Adlercreutzia、Collinsella、Klebsiella、Parabacteroides和Sutterella相对丰度提高,同时,便秘组Bacteroides和Odoribacter相对丰度降低,这20 个关键菌属中大部分是对肠道健康有益的菌属,但是也有病原菌,其中,Bacteroides、Faecalibacterium、Blautia是短链脂肪酸的产生菌属,Faecalibacterium与抗炎相关,Collinsella与促炎作用相关,Parabacteroides和Klesbsiella是能够引起腹腔内膜感染、腹泻等病症的相关致病菌,Blautia可将肠道中气体转化成乙酸,使气体得以清除[31]。这提示益生菌剂能够通过调节关键菌属丰度比例来调整肠道微生物菌群结构,可能与菌属本身及其代谢产物的抗炎、促炎作用具有密切的联系,同时也说明菌群的多样性与受试人群本身病理状态有很大的相关性。
本研究对受试人群种水平的变化进行了探究发现,检测出的肠道菌群大多属于拟杆菌型,这与Arumugam等[32]提出的肠型之说相符;Wang Tingting等[33]采用454焦磷酸测序技术对肠道菌群16S rRNA V3分析也确定健康人群肠道中富集了普通拟杆菌和单形拟杆菌。便秘组Akkermansia muciniphila、Eubacterium biforme在第2周,Bacteroides fragilis、Faecalibacterium prausnitzii在第5周至停止,Ruminococcus gnavus在第4周及停止干预后的第2周均在益生菌剂的干预下出现,且含量分别高达0.42%、0.02%、16.17%、2.99%、0.08%,Bacteroides ovatus和Prevotella stercorea则随干预时长降低。其中,Akkermansia muciniphila被认为是健康人体肠道中最丰富的黏液溶解细菌,其数目和乙酸盐、丁酸盐含量之间存在很强的相关性,在降解黏蛋白的同时会释放乙酸、丁酸等短链脂肪酸,肠道中低水平的Akkermansia muciniphila可能导致黏膜层的变薄,从而导致肠道屏障功能减弱,使肠道内的毒素更容易侵入人体[34-35]。Bacteroides fragilis是人体结肠中的一个优势菌,它通过产生独特的荚膜多糖A改变其表面抗原性,当其含量过低时会引起肠道炎症[36]。Faecalibacterium prausnitzii是肠道共生菌,是硬壁菌门的主要成员,能通过产丁酸盐为肠道提供能量并发挥抗炎作用[37]。Bacteroides ovatus能够降解人体不能代谢的植物多糖,为宿主提供来自食物的10%~15%的能量[38]。腹泻组的Bifidobacterium longum、Bacteroides stercoris在第6周(9.09%、0.33%)、Actinobacillus parahaemolyticus在第1周(0.12%)、Anoxybacillus kestanbolensis在第4周(0.07%)、Lactobacillus brevis、Blautia producta及Eubacterium dolichum在停止干预后(0.01%、0.03%、0.09%)均出现。对于其他组而言,在益生菌剂干预下,腹胀受试患者(A1_S6)的瘤胃乳酸杆菌(Lactobacillus ruminis)在益生菌剂干预第6周(0.12%)、肠易激综合征患者(I1_S1、I1_S2、I1_S4)及排便不规律患者(N1_S6)的Bacteroides coprophilus在第1~6周时开始出现,这提示益生菌剂具有短期调控菌群多样性及相对丰度的作用,并且通过菌群的代谢产物的有益作用达到益生功能。由于这些菌种含量过低不易检测,再加上益生菌剂对于单个菌种的持续作用并不明显,再次印证了益生菌剂干预下种水平的不显著变化可能与受试人群的病理状态有关。此外,对一些机制不明的菌有待进一步研究。
本研究通过对不同健康状况受试人群施于益生菌剂的干预,采集粪便,利用Ion Torrent PGM测序技术和多变量统计学方法分析得知,益生菌剂对受试人群肠道微生物多样性均有不同程度的调整作用。益生菌剂能够降低便秘人群Bacteroidetes、Rikenellaceae,升高Firmicutes、Alcaligenaceae、Coriobacteriaceae的相对丰度,提高腹泻人群Proteobacteria、Alcaligenaceae、Lachnospiraceae、Erysipelotrichaceae并降低Veillonellaceae,提高健康状况人群Proteobacteria、Actinobacteria、Enterobacteriaceae、Bifidobacteriaceae、Rikenellaceae、Odoribacteraceae、Desulfovibrionaceae,使其相对丰度向健康人群靠近。并且,益生菌剂在属水平上对腹泻组的调整效果优于其他两组。重要的是,Akkermansia muciniphila、Bacteroides fragilis及Faecalibacterium prausnitzii等的相对丰度有不同程度的提高,这些菌的出现将对维护宿主健康具有价值。因此本研究认为,益生菌剂能起到改变肠道微生物多样性的功效,可以通过调整关键菌群间的丰度比例起到抑制肠道有害微生物生长、促进部分有益菌的增殖并维持肠道菌群结构稳态的作用,进一步明确了利用益生菌剂调节和恢复便秘、腹泻人群肠道健康具有科学价值。
参考文献:
[1] BÄCKHED F, LEY R E, SONNENBURG J L, et al. Host-bacterial mutualism in the human intestine[J]. Science, 2005, 307: 1915-1920.DOI:10.1126/science.1104816.
[2] 胡旭, 王涛, 王沥, 等. 肠道共生微生物与健康和疾病[J]. 中国微生态学杂志, 2012, 24(12): 1134-1139. DOI:10.13381/j.cnki.cjm.2012.12.004.
[3] 林璋, 祖先鹏, 谢海胜, 等. 肠道菌群与人体疾病发病机制的研究进展[J].药学学报, 2016, 51(6): 843-852. DOI:10.16438/j.0513-4870.2015-0803.
[4] JANSSEN A W F, KERSTEN S. The role of the gut microbiota in metabolic health[J]. FASEB Journal, 2015, 29(8): 3111-3123.DOI:10.1096/fj.14-269514.
[5] ECKBURG P B, BIK E M, BERNSTEIN C N, et al. Diversity of the human intestinal microbial flora[J]. Science, 2005, 308: 1635-1638.DOI:10.1126/science.1110591.
[6] BIBEL D J. Elie Metchnikoff’s bacillus of long life[J]. ASM News,1988, 54: 661-665.
[7] CHEN C C, KONG M S, LAI M W, et al. Probiotics have clinical,microbiologic, and immunologic efficacy in acute infectious diarrhea[J].The Pediatric Infectious Disease Journal, 2010, 29(2): 135-138.
[8] HEO J, KIM S K, PARK K S, et al. A double-blind, randomized,active drug comparative, parallel-group, multi-center clinical study to evaluate the safety and efficacy of probiotics (Bacillus licheniformis,Zhengchangsheng®capsule) in patients with diarrhea[J]. Intestinal Research, 2014, 12(3): 236-244. DOI:10.5217/ir.2014.12.3.236.
[9] GUANDALINI S, MAGAZZU G, CHIARO A, et al. VSL#3 improves symptoms in children with irritable bowel syndrome: a multicenter,randomized, placebo-controlled, double-blind, crossover study[J].Journal of Pediatric Gastroenterology & Nutrition, 2010, 51(1): 24-30.DOI:10.1097/MPG.0b013e3181ca4d95.
[10] 丁圣, 蒋菁莉, 刘松玲, 等. 长双歧杆菌BBMN68对便秘模型小鼠的通便作用[J]. 食品科学, 2011, 32(3): 195-198.
[11] 张敏, 张艳, 兰国伟, 等. 高通量测序技术在乳制品微生物多样性中的研究进展[J]. 中国农学通报, 2016, 32(32): 48-52.
[12] 李春慧, 蒲万霞, 吴润, 等. 微生态制剂应用研究概况[J]. 动物医学进展, 2013(11): 114-118. DOI:10.3969/j.issn.1007-5038.2013.11.027.
[13] 臧凯丽, 江岩, 孙勇, 等. 微生态制剂调节便秘、腹泻人群肠道菌群结构与产短链脂肪酸关键菌属的相关性[J]. 食品科学, 2018, 39(5):155-165. DOI:10.7506/spkx1002-6630-201805024.
[14] 张烽, 张晨虹. 膳食营养与肠道微生物组[J]. 生命科学, 2017(7):695-706. DOI:10.13376/j.cbls/2017094.
[15] QIN J, LI R, RAES J, et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010, 464: 59-65.DOI:10.1038/nature08821.
[16] 刘开朗, 王加启, 卜登攀, 等. 人体肠道微生物多样性和功能研究进展[J]. 生态学报, 2009, 29(5): 2589-2594. DOI:10.3321/j.issn:1000-0933.2009.05.048.
[17] MURPHY E F, COTTER P D, HEALY S, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet,obesity and time in mouse models[J]. Gut, 2010, 59(12): 1635-1642.DOI:10.1136/gut.2010.215665.
[18] 王秀. 微生态制剂对肠道菌群紊乱的调节作用[J]. 江西饲料,2012(5): 15-16. DOI:10.3969/j.issn.1008-6137.2012.05.006.
[19] 张家超, 郭壮, 孙志宏, 等. 益生菌对肠道菌群的影响: 以Lactobacillus casei Zhang研究为例[J]. 中国食品学报, 2011, 11(9):58-68. DOI:10.3969/j.issn.1009-7848.2011.09.007.
[20] 郭慧慧, 黄帅, 王璐璐, 等. 肠道菌群对机体营养物质的代谢研究[J].中国医药生物技术, 2016, 11(4): 340-345. DOI:10.3969/j.issn.1673-713X.2016.04.010.
[21] SEGATA N, HAAKE S K, MANNON P, et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples[J]. Genome Biology, 2012,13(6): 1-18. DOI:10.1186/gb-2012-13-6-r42.
[22] WANG Z Q, AMMAR E M, ZHANG A, et al. Engineering Propionibacterium freudenreichii, subsp. shermanii, for enhanced propionic acid fermentation: effects of overexpressing propionyl-CoA:succinate CoA transferase[J]. Metabolic Engineering, 2015, 27: 46-56.DOI:10.1016/j.ymben.2014.10.005.
[23] JANG Y S, IM J A, CHOI S Y, et al. Metabolic engineering of Clostridium acetobutylicum for butyric acid production with high butyric acid selectivity[J]. Metabolic Engineering, 2014, 23: 165-174.DOI:10.1016/j.ymben.2014.03.004.
[24] DEN BESTEN G, VAN EUNEN K, GROEN A K, et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota,and host energy metabolism[J]. Journal of Lipid Research, 2013,54(9): 2325-2340.
[25] REMELY M, AUMUELLER E, MEROLD C, et al. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity[J]. Gene, 2014, 537(1): 85-92.DOI:10.1016/j.gene.2013.11.081.
[26] LAYDEN B T, ANGUEIRA A R, BRODSKY M, et al. Short chain fatty acids and their receptors: new metabolic targets[J]. Translational Research the Journal of Laboratory & Clinical Medicine, 2013, 161(3):131-140. DOI:10.1016/j.trsl.2012.10.007.
[27] CARMODY R N, GERBER G K, LUEVANO J M, et al. Diet dominates host genotype in shaping the murine gut microbiota[J].Cell Host & Microbe, 2015, 17(1): 72-84. DOI:10.1016/j.chom.2014.11.010.
[28] ZHANG Chenhong, ZHANG Menghui, PANG Xiaoyan, et al.Structural resilience of the gut microbiota in adult mice under highfat dietary perturbations[J]. ISME Journal, 2012, 6(10): 1848-1857.DOI:10.1038/ismej.2012.27.
[29] 朱庆超. 高脂状态下肠道微生态异常与大肠癌发病风险的相关实验研究[D]. 上海: 上海交通大学, 2014: 148-169.
[30] 明珠, 陈庆森, 刘雪姬, 等. 乳源酪蛋白糖巨肽对溃疡性结肠炎小鼠肠道菌群多样性的影响[J]. 食品科学, 2016, 37(5): 154-161.DOI:10.7506/spkx1002-6630-201605028.
[31] CLAVEL T, GOMES-NETO J C, LAGKOUVARDOS I, et al.Deciphering interactions between the gut microbiota and the immune system via microbial cultivation and minimal microbiomes[J].Immunological Reviews, 2017, 279(1): 8-22. DOI:10.1111/imr.12578.
[32] ARUMUGAM M, RAES J, PELLETIER E, et al. Addendum:enterotypes of the human gut microbiome[J]. Nature, 2011, 473: 174-180. DOI:10.1038/nature13075.
[33] WANG Tingting, CAI Guoxiang, QIU Yunping, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers[J]. The ISME Journal, 2012, 6: 320-329.DOI:10.1038/ismej.2011.109.
[34] DAO M C, EVERARD A, ARONWISNEWSKY J, et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology[J]. Gut,2016, 65(3): 426-436. DOI:10.1136/gutjnl-2014-308778.
[35] BRAHE L K, CHATELIER E L, PRIFTI E, et al. Specific gut microbiota features and metabolic markers in postmenopausal women with obesity[J]. Nutrition & Diabetes, 2015, 5(6): 1-7. DOI:10.1038/nutd.2015.9.
[36] 冯淑贞, 张和平. 脆弱拟杆菌的研究进展[J]. 微生物学通报, 2015,42(7): 1366-1371. DOI:10.13344/j.microbiol.china.140778.
[37] LU H F, WU Z W, XU W, et al. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection: intestinal microbiota of HBV cirrhotic patients[J]. Microbial Ecology, 2011, 61(3): 693-703.DOI:10.1007/s00248-010-9801-8.
[38] GRIFFIN J L, WANG X, STANLEY E. Does our gut microbiome predict cardiovascular risk? a review of the evidence from metabolomics[J]. Circulation Cardiovascular Genetics, 2015, 8(1):187-191. DOI:10.1161/CIRCGENETICS.114.000219.
Probiotics Modulate the Structure and Abundance of Gut Mircrobiota in Populations with Intestinal Diseases
ZANG Kaili1, JIANG Yan1, SUN Yong2,*, YAN Yali1,*, CHEN Qingsen1, ZHAO Linsen3, ZHAO Pei1, CUI Wenjing3, MA Xinying3
(1. Tianjin Key Laboratory of Food Biotechnology, College of Biotechnology and Food Science, Tianjin University of Commerce,Tianjin 300134, China; 2. Beijing Academy of Food Sciences, China Meat Research Center, Beijing 100068, China;3. Hebei Inatural Biological Technical Company, Shijiazhuang 050800, China)
Abstract:The aim of the present study was to explore the regulatory effect of probiotics on the intestinal microbial diversity and abundance in patient populations with intestinal diseases. A probiotic preparation (PPr) was regularly consumed at a constant amount for 6 weeks by healthy, constipated, diarrheal, abdominal bloating, irritable bowel syndrome and irregular defecation populations. Before, during and after the experiment, fecal samples were collected for bacterial genomic DNA extraction. The sequencing of 16S rRNA V3 region was performed with the Personal Genome Machine (PGM, Ion Torrent).The sequencing data were used for diversity analysis by bioinformatics and multivariate statistical analysis. When the sequencing depth was satisfactory, the effect of the probiotics in correcting gut microbiota imbalance in the subjects was evaluated at the four levels of phylum, family, genus and even species. All sequencing reads were divided into 2 320 operational taxonomic units at a 97% similarity level. Bacteroidestes, Firmicutes, Proteobacteria and Actinobacteria were the dominant phyla, accounting for 99.81% of the total number of sequences. For all the subjects, the probiotics had a longlasting positive impact on Lachnospiraceae whereas Alcaligenaceae, Rikenellaceae, Bifidobacteria (Bifidobacteriaceae) and other dominant bacteria returned to the pre-intervention level when the intervention was withdrawn. The effects of probiotics on gut microbiota were different among all patient populations, and both Cyanobacteria and Fusobacteria were detected in the constipation group after consumption of the priobiotics. Principal component analysis (PCA) analysis showed that the regulatory effect of the probiotics in the diarrheal group was superior to that in two other groups during the first to the fourth week of intervention. The diversity and abundance of 10 genera including Faecalibacterium, Acinetobacter, Blautia,Clostridium, Dialister, Eggerthella, Granulicatella, Lactobacillus, Oxalobacter, Pyramidobacter were significantly increased in the diarrheal group, while only three genera: Prevotella, Megamonas and Collinsella in the constipation group changed. Moreover,the other groups exhibited a change in the abundance of Adlercreutzia, Collinsella, Klebsiella, Parabacteroides, and Sutterella.PCA analysis also revealed obvious differences in the regulatory effect of the probiotics among different gut microbial populations and that the probiotics had different effects in improving and treating different intestinal diseases. In addition, Bacteroides and Odoribacter in the constipation group tended to decrease to normal levels. Heatmap analysis showed that several key functional fungi, such as Akkermansia muciniphila, Bacteroides fragilis and Faecalibacterium prausnitzii, were found to appear only at some stages, but disappear after the intervention was withdrawn. At this time, the original Bacteroides ovatus began to decrease. The appearance of these bacteria was beneficial to host health. In summary, probiotic intervention can play a role in altering the diversity and abundance of gut microbes and inhibit the growth of harmful microorganisms in the gut. More importantly, the appearance of beneficial bacteria manifests the physiological effectiveness of the probiotics. Therefore, the probiotics have promising applications in maintaining a healthy state of intestinal microflora.
Keywords:probiotics; intestinal disease; flora abundance; Ion Torrent PGM sequencing technology; bioinformatics
DOI:10.7506/spkx1002-6630-201813021
收稿日期:2018-01-03
基金项目:国家自然科学基金面上项目(31071522)
第一作者简介:臧凯丽(1993—),女,硕士研究生,研究方向为生物活性物质的研究与开发。E-mail:1623249305@qq.com
*通信作者简介:孙勇(1977—),男,副研究员,博士,研究方向为食品营养化学。E-mail:sy183509@163.com闫亚丽(1962—),女,副教授,硕士,研究方向为发酵生物技术、微生物学。E-mail:yyali@tjcu.edu.cn
中图分类号:TS201.3
文献标志码:A
文章编号:1002-6630(2018)13-0133-11
引文格式:臧凯丽, 江岩, 孙勇, 等. 益生菌剂调整肠道疾病人群菌群结构丰度水平的研究[J]. 食品科学, 2018, 39(13): 133-143.
DOI:10.7506/spkx1002-6630-201813021. http://www.spkx.net.cn
ZANG Kaili, JIANG Yan, SUN Yong, et al. Probiotics modulate the structure and abundance of gut mircrobiota in populations with intestinal diseases[J]. Food Science, 2018, 39(13): 133-143. (in Chinese with English abstract)
DOI:10.7506/spkx1002-6630-201813021. http://www.spkx.net.cn