表1 引物序列
Table 1 Primer sequences used in this study
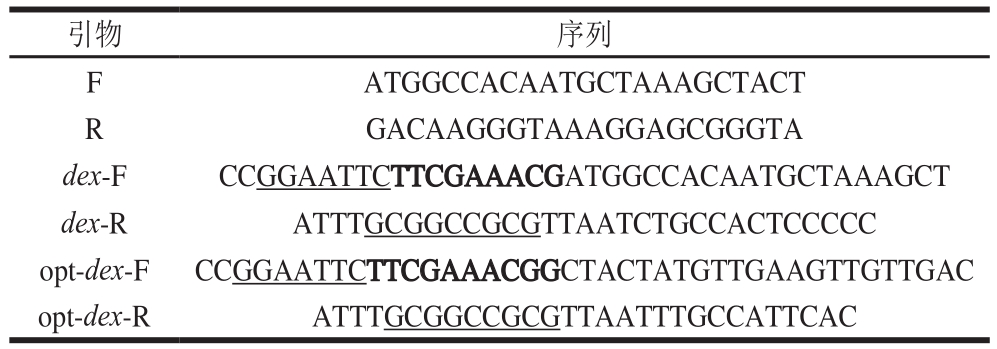
注:下划线为EcoRI及NotI酶切位点;加粗部分为kozark序列。
吴 敏,张宇馨,胡雪芹,张洪斌*
(合肥工业大学生物与医学工程学院,安徽 合肥 230009)
摘 要: 以右旋糖酐酶产生菌棘孢青霉F1001基因组为模板反转录合成右旋糖酐酶的cDNA(dex),基因全长1 866 bp,根据毕赤酵母密码子偏好性优化dex序列获得优化右旋糖酐酶基因(opt-dex),分别构建dex-pPICZαA和opt-dex-pPICZαA重组质粒,电击转入毕赤酵母X33中构建重组子。通过蓝色右旋糖酐T-2000平板以及摇瓶发酵筛选获得产右旋糖酐酶的重组酵母菌株。重组酶的酶学性质分析显示,重组酶分子质量65 kDa、最适pH 5.0、最适温度35 ℃,专一作用于α-1,6糖苷键。在摇瓶水平上对重组毕赤酵母表达条件进行优化,优化培养条件为培养温度25 ℃、初始pH 5.0、每24 h甲醇添加量1%(体积分数)、每24 h山梨醇添加量5 g/L、吐温-80添加量4 g/L、摇瓶装液量50 mL/500 mL锥形瓶,优化后的重组右旋糖酐酶分泌表达酶活力提高到240.74 U/mL。重组酵母X33是一株适合外源表达棘孢青霉右旋糖酐酶基因的工程菌,该重组酶可替代棘孢青霉右旋糖酐酶直接应用于工业生产催化制备右旋糖酐。
关键词: 右旋糖酐酶;基因优化;毕赤酵母;表达
右旋糖酐酶是一种能专一性断裂右旋糖酐α-1,6糖苷键的水解酶,主要催化产物包括低聚右旋糖酐以及低聚异麦芽糖等。低聚右旋糖酐T70、T40、T20是重要的血浆代用品,具有补充血容量、改善微循环等作用 [1] 。低聚异麦芽糖是一种功能性低聚糖,具有调节肠道菌群、改善人体内微生态环境的作用 [2-3] 。除此之外,右旋糖酐酶及其衍生物还可用于糖蜜制作和饮料加工 [4] 、防治龋齿、保持口腔卫生 [5-6] 等,具有重要的应用价值。目前关于右旋糖酐酶的研究主要集中在产生菌株的挖掘以及酶学性质的研究,但野生菌株遗传背景不清晰、代谢复杂等缺陷 [7] ,极大程度限制了右旋糖酐酶在工业生产中的应用。为解决这一问题,国内外学者开始在工程菌中对右旋糖酐酶基因进行克隆表达,其中毕赤酵母以其绿色安全、表达量高的优势受到广泛关注。1996年Roca等 [8] 首次在毕赤酵母菌株中表达了朱黄青霉右旋糖酐酶(Penicillium minioluteum IMI068219),甲醇诱导106 h后右旋糖酐酶产量达到3.2 g/L,酶活力为65.3 U/mg。梁达奉 [9] 分别构建了诱导型、组成型2 种不同的重组酵母菌分泌表达朱黄青霉右旋糖酐酶(Penicillium minioluteum HI-4),摇瓶酶活力分别为75、60.4 U/mL。黄曾慰等 [10] 通过密码子优化进一步提高朱黄青霉右旋糖酐酶(Penicillium minioluteum HI-4)在毕赤酵母中异源表达产量。青霉菌是右旋糖酐酶的主要来源菌属,包括绳状青霉 [11] 、点青霉 [12-13] 、嗜松青霉 [14] 、朱黄青霉、棘孢青霉 [15-16] 等。但目前关于青霉菌属右旋糖酐酶的克隆表达主要集中于朱黄青霉,其他青霉属右旋糖酐酶的相关研究鲜有报道,棘孢青霉F1001 [16] 是本实验室筛选获得的高产右旋糖酐酶菌株,具有较高的产酶能力,所分泌表达的右旋糖酐酶是内切型右旋糖酐酶并具有优良的酶学性质,对其进行进一步的克隆表达分析有利于加强对青霉属右旋糖酐酶的认识,也有利于提高右旋糖酐酶的工业应用潜力。
本实验合成了棘孢青霉F1001右旋糖酐酶基因(GenBank登录号KF999646.1)并对序列进行优化,分别将优化前后的右旋糖酐酶基因序列克隆至毕赤酵母中进行异源表达,特异性筛选获得阳性菌株。对重组酶的酶学性质进行考察并对摇瓶发酵条件进行优化探索。
棘孢青霉H5、大肠杆菌DH5α为本实验室保存;毕赤酵母X33、质粒pPICZαA 美国Invitrogen公司;限制性内切酶、连接酶、聚合酶 北京全式金生物有限公司;Trizol提取试剂盒、第1链cDNA合成试剂盒、博来霉素 生工生物工程上海(股份)有限公司。
LLB培养基:胰蛋白胨10 g/L、酵母浸粉5 g/L、NaCl 5 g/L,pH 7.0。棘孢青霉诱导培养基:右旋糖酐T70 15 g/L、蛋白胨5 g/L、K 2 PO 4 ·3H 2 O 1 g/L、FeSO 4 ·7 H 2 O 0.01 g/L、KCl 0.5 g/L、MgSO 4 ·7 H 2 O 0.5 g/L,pH 5.5。BMGY培养基:酵母浸出物10 g/L、蛋白胨20 g/L、100 mmol/L磷酸钠(pH 5.0)、无氨基酵母氮源YNB 13.4 g/L、生物素0.4 mg/L、甘油10 g/L。BMMY培养基:酵母浸出物10 g/L、蛋白胨20 g/L、100 mmol/L磷酸钠(pH 5.0)、YNB 13.4 g/L、生物素0.4 mg/L、体积分数1%甲醇。
蓝色右旋糖酐T-2000平板:蓝色右旋糖酐20 g/L、YNB 13.4 g/L、生物素0.4 mg/L,在倒置的皿盖上加入1%(相对于固体培养基体积)甲醇。
1.3.1 菌株培养
棘孢青霉培养:PDA平板连续活化棘孢青霉菌株,单菌落接种至棘孢青霉诱导培养基中(50 mL/250 mL锥形瓶),30 ℃,250 r/min培养5 d。
大肠杆菌培养:将大肠杆菌接入50 mL LLB培养基中于37 ℃、220 r/min培养24 h。
毕赤酵母培养:将重组菌单菌落接入30 mL YPD培养基中,30 ℃、250 r/min培养过夜至OD 600 nm 值为5,然后以体积分数1%接种量转接至50 mL BMGY培养基中去葡萄糖抑制,30 ℃、250 r/min培养至OD 600 nm 值为4~6时,离心,BMMY重悬菌体,25 ℃、250 r/min培养,每24 h添加甲醇至终体积分数1%诱导产酶。
1.3.2 酶活力和总蛋白的测定
右旋糖酐酶活力测定采用还原糖测定方法——DNS法 [17] :发酵液于4 ℃、8 000 r/min离心5 min,弃沉淀,上清液稀释一定倍数。将4 mL 0.02 mol/L pH 5.0的醋酸盐缓冲液配制的3%右旋糖酐T70溶液置于35 ℃保温10 min后加入1 mL稀释的酶液,保温1 h后,取样500 μL,与375 μL DNS混合,沸水浴5 min,冷却至室温定容至终体积5.375 mL。灭活的酶液作为对照组,每分钟产生1 μmol还原糖所需的酶量定义为一个酶活力单位(U)。
总蛋白测定:利用Bradford法 [18] 测定蛋白含量,以牛血清蛋白为标准绘制蛋白标准曲线。将1 mL发酵液与4 mL考马斯亮蓝G250在常温下反应10 min后,在595 nm波长下测定吸光度,计算蛋白含量。
1.3.3 dex基因合成
活化培养棘孢青霉菌,离心收集菌体,无菌水洗涤,液氮研磨,试剂盒提取总RNA,反转录合成第1链DNA,根据NCBI数据库中已知的右旋糖酐酶基因序列进行分析,以朱黄青霉HI-4(GenBank号L41562.1)编码序列为模板,设计特异性上游引物F、下游引物R(表1),以第1链DNA为模板进行聚合酶链式反应(polymerase chain reaction,PCR)特异性扩增,得到双链DNA片段,即dex基因序列,通过T克隆与pMD18-T载体连接,热击转入E. coli DH5α中,蓝白斑筛选获得阳性克隆子,测序获得棘孢青霉右旋糖酐酶基因序列,并将序列提交至GenBank。
表1 引物序列
Table 1 Primer sequences used in this study
注:下划线为EcoRI及NotI酶切位点;加粗部分为kozark序列。
1.3.4 右旋糖酐酶密码子优化
利用软件Gene Designer对棘孢青霉右旋糖酐酶基因dex进行密码子优化,通过同义密码子替换的方式调整目的基因GC相对含量以及密码子偏好指数,序列文件送至生工生物工程(上海)股份有限公司进行全基因合成。
1.3.5 重组表达质粒构建
根据右旋糖酐酶序列文件设计合成引物(表1),分别在上下游引物中添加EcoRI以及NotI位点(下划线表示),并在上游引物中引入Kozark序列(加粗部分)。PCR分别扩增dex、opt-dex序列后通过双酶切的方式与载体pPICZαA连接,42 ℃热击将连接产物转入E. coli DH5α中,在含有博来霉素抗性的LLB平板上筛选重组质粒,PCR以及双酶切鉴定阳性克隆并测序。
1.3.6 重组毕赤酵母构建及表达
SacI线性化重组质粒后转入毕赤酵母X33中,通过高质量浓度博来霉素(100、200、300、400、500 μg/mL)连续筛选,然后将筛选获得的单克隆点种在蓝色右旋糖酐T-2000平板进行特异性筛选,阳性菌落能够分泌产生右旋糖酐酶水解蓝色右旋糖酐T-2000,在单菌落周围形成透明圈。选取能产生水解圈的单克隆进行摇瓶表达,并根据1.3.2节测定诱导发酵96 h的重组酶活力,筛选产酶能力高的重组酵母菌株。
1.3.7 重组右旋糖酐酶分离纯化
将500 mL发酵粗酶液与500 mL冰预冷丙酮混合,4 ℃静置10 min,8 000 r/min离心15 min弃上清液,用冰预冷丙酮洗涤3 次后,取10 mL 0.02 mol/L pH 5.0醋酸盐缓冲液溶解沉淀,粗酶液浓缩50 倍。将10 mL浓缩液与磁珠混合,在磁场作用下实现目标蛋白与镍离子的结合、杂蛋白洗脱以及目标蛋白的分离,获得较高纯度的重组右旋糖酐酶,十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)检验纯化效果。
1.3.8 重组右旋糖酐酶酶学性质测定
在15~70 ℃分别测定重组右旋糖酐酶活力,考察重组酶的最适反应温度,并在梯度温度下保温1 h考察重组酶热稳定性,以酶活力最高为100%,计算相对酶活力;配制pH 2~9的缓冲液,测定不同pH值环境中重组右旋糖酐酶活力,考察重组酶最适反应pH值,并在梯度pH值条件下孵育1 h后考察重组酶的pH值耐受性,以酶活力最高为100%,计算相对酶活力。
选择含有不同键型的多糖,测定不同底物催化条件下的重组酶活力,考察重组右旋糖酐酶的底物特异性。然后选择重组右旋糖酐酶的最适反应底物,配制成不同浓度的底物溶液,测定重组右旋糖酐酶反应初速率,计算得到重组酶的米氏常数K m 值和最大反应速率v max 。
1.3.9 摇瓶优化单因素试验
为进一步提高毕赤酵母分泌表达外源蛋白能力,对毕赤酵母摇瓶发酵条件进行优化,基本培养条件同1.3.1节。考察每24 h添加甲醇至终体积分数为0.5%、1%、1.5%、2%、2.5%、3.0%时重组酵母产酶能力;考察摇瓶装液量为25、50、100、150、200 mL/500 mL时重组酵母产酶能力;考察培养基pH 3.0、4.0、5.0、6.0、7.0、8.0时重组菌产酶能力;考察诱导温度为20、25、30 ℃时重组酵母菌产酶能力;接着优化非离子表面活性剂吐温-80、吐温-20及其不同添加量对重组酵母产酶影响;优化碳源种类山梨醇、甘油及其添加量对重组菌产酶能力的影响。实验均做3 个平行,标准差在图中以误差线标示。
提取棘孢青霉总RNA,如图1A所示,获得28S、18S的rRNA条带,无明显拖尾现象,说明总RNA质量良好。再以总RNA中的mRNA为模板反转录合成cDNA,PCR扩增得到右旋糖酐酶基因dex,如图1B所示,基因长度为1 866 bp。

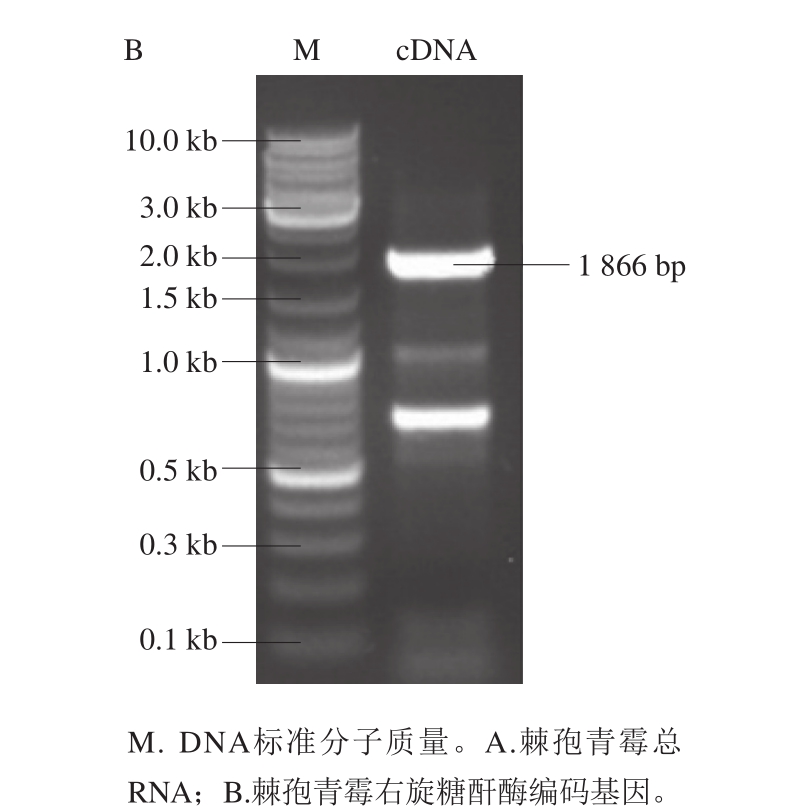
图1 棘孢青霉右旋糖酐酶基因电泳图
Fig. 1 Agarose gel electrophoresis of the Penicillium aculeatum gene encoding dextranase
表2 棘孢青霉右旋糖酐酶基因优化前后密码子使用频率
Table 2 Codon usage frequency of dex and opt-dex in Pichia pastoris
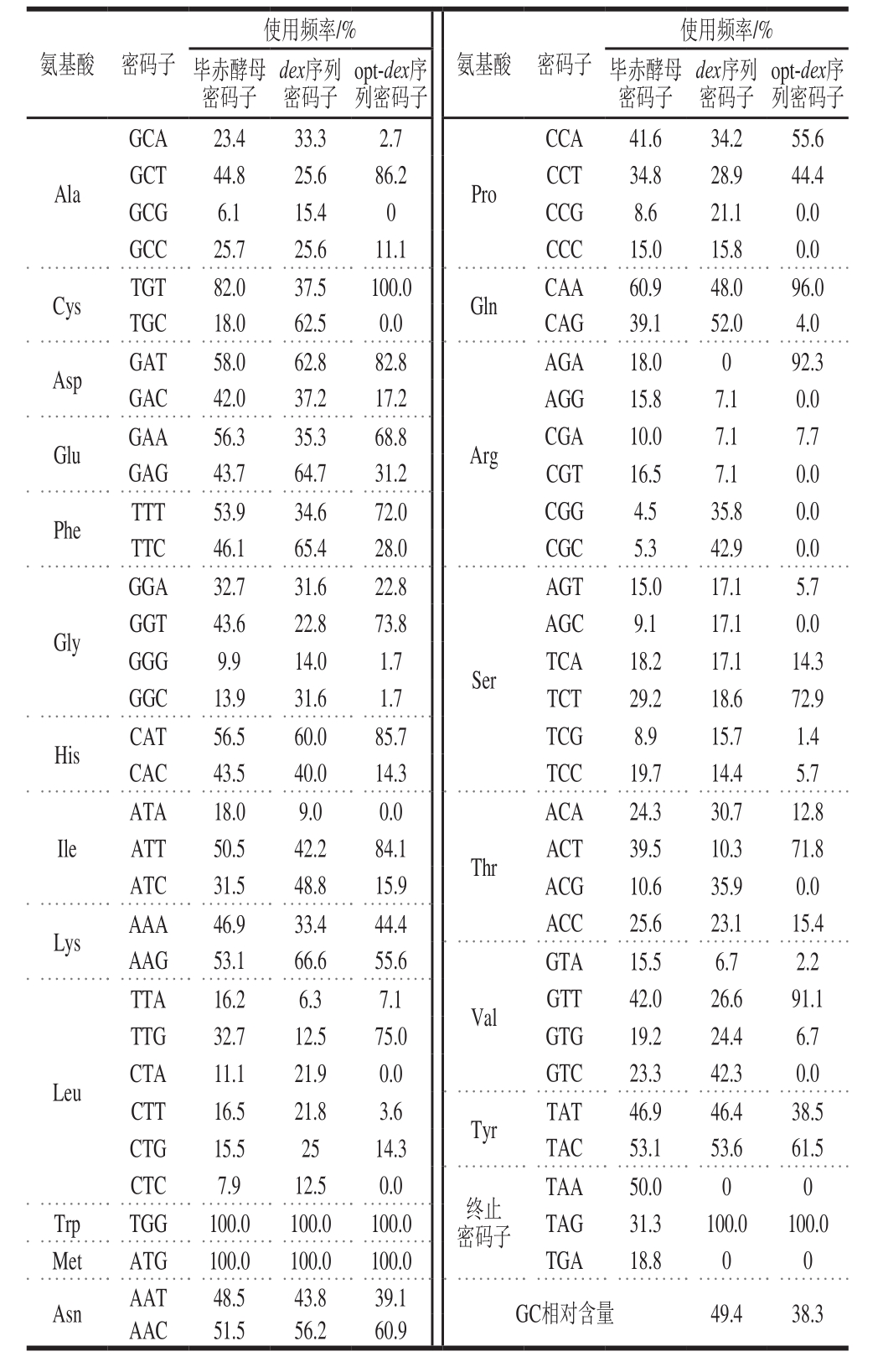
通过同义突变,将在毕赤酵母中利用的低频密码子替换成高频密码子,右旋糖酐酶基因序列优化前后密码子分布如表2所示,例如编码Ala的GCG密码子在毕赤酵母中使用频率极低,仅6.1%,因此优化后的右旋糖酐酶序列中不再使用GCG编码Ala,采用酵母偏好性较高的GCT进行编码。再综合考虑基因序列的GC相对含量、酶切位点、不稳定序列等因素,优化后的基因序列的密码子偏好指数从原始序列的0.65调整至0.92(密码子偏好指数在0.8~1.0之间更有利于外源蛋白的表达),序列GC相对含量从49.4%降至38.3%,去除了原始序列中的TAAAT不稳定序列。
分别将dex与opt-dex基因与表达质粒载体pPICZαA连接,SacI线性化后电击转化至毕赤酵母X33基因组中,蓝色右旋糖酐平板筛选结果如图2所示,图2A、2B分别表示密码优化后opt-dex以及密码子优化前dex序列在毕赤酵母中的表达情况。并且图2A显示甲醇诱导24 h后的情况,图2B表示甲醇诱导72 h后才出现微弱的水解圈,可以初步判断密码子opt-dex序列在毕赤酵母中具有更高的右旋糖酐酶表达能力。
进一步选择图2A中水解圈较明显的A1、A3、A5、A6、A7、A10以及图2B中唯一的阳性菌落B13进行摇瓶表达,结果如图3所示,B13的表达能力明显低于A组菌株。其中A组中A5具有较高产酶活力,在摇瓶的表达酶活力达到89.56 U/mL,选择该菌株进行后续研究。

图2 阳性克隆子筛选
Fig. 2 Selection of positive transformants

图3 转化子的摇瓶发酵筛选
Fig. 3 Selection of transformants producing dextranase by shake flask fermentation
发酵粗酶液经浓缩后如图4A所示,样品中含有大量的杂蛋白条带以及其他杂质成分,以至于对其他泳道造成了严重的挤压。磁珠纯化后的重组右旋糖酐酶如图4B所示,目标条带单一,说明纯化后的酶液具有较高纯度。SDS-PAGE同时也表明重组右旋糖酐酶大小为65 kDa,与理论蛋白大小符合。
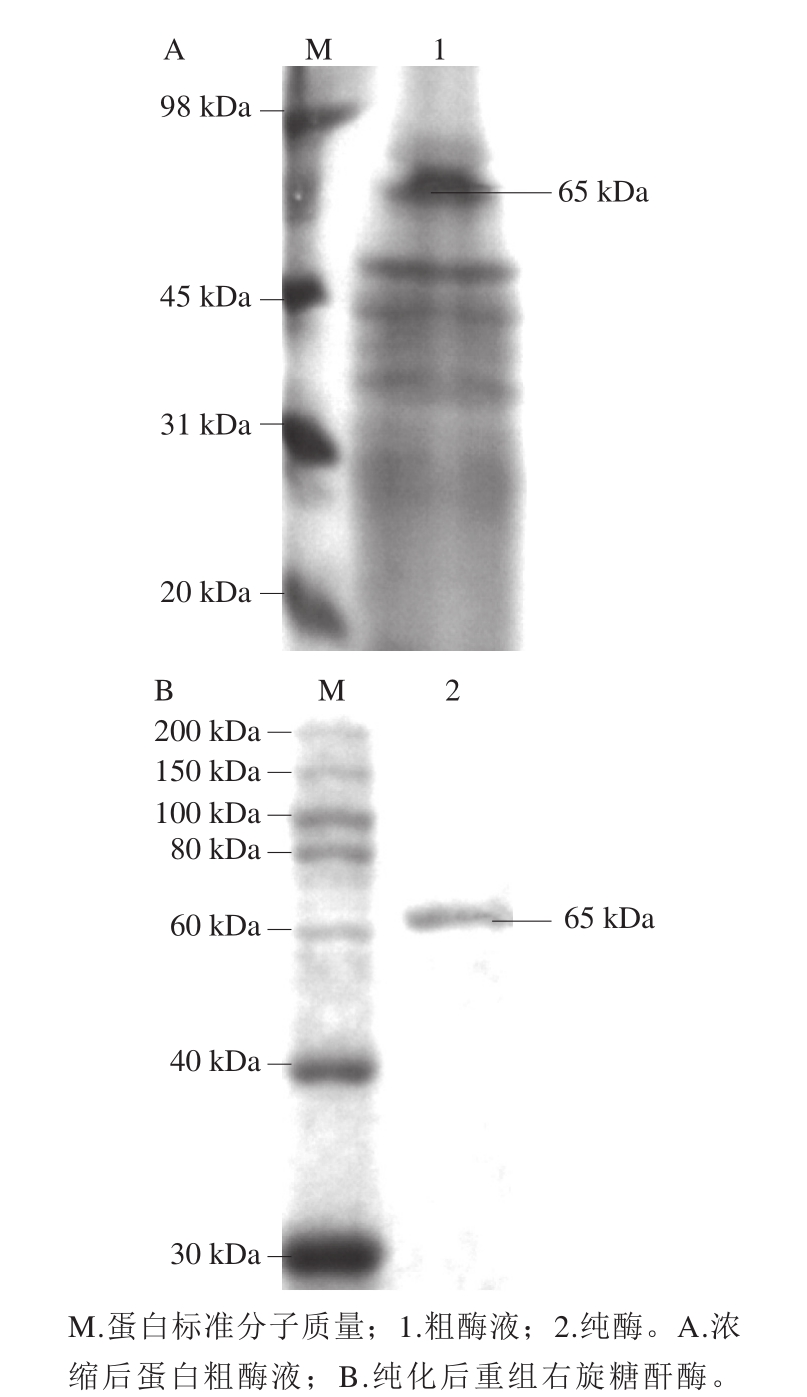
图4 重组右旋糖酐酶纯化结果
Fig. 4 SDS-PAGE analysis of purified recombinant dextranase
2.5.1 重组右旋糖酐酶最适催化条件及稳定性
如图5A所示,重组右旋糖酐酶最适催化温度为35 ℃,随着温度升高酶活力逐渐降低,但在70 ℃的高温条件下进行催化反应仍然能够表现出40%的剩余酶活力。与天然棘孢青霉右旋糖酐酶相比较 [16] ,具有相同的最适催化温度,以及相似的对高温和低温的酶活力响应趋势。但是在温度稳定性实验中,天然酶在45 ℃中保存1 h仍然保留95.67%的酶活力,而重组酶在45 ℃保温1 h剩余酶活力仅为75%,并且在50 ℃保温1 h后重组右旋糖酐酶剩余酶活力降低至46%,而天然酶剩余活力在80%以上 [16] 。说明经过酵母表达的重组右旋糖酐酶在温度稳定性上与天然酶相比有一定程度的降低。
对重组右旋糖酐酶的最适催化pH值考察,结果如图5B所示,发现其最适催化pH值为5.0,当pH值大于5.0时,酶活力迅速降低,当pH值升高到9.0时,重组右旋糖酐酶几乎全部失活。重组右旋糖酐酶在pH 4.0~7.0缓冲液中孵育1 h后能够保留80%以上的酶活力。重组酶的最适催化pH值以及在不同pH值条件下的稳定性与天然棘孢青霉右旋糖酐酶一致 [16] 。
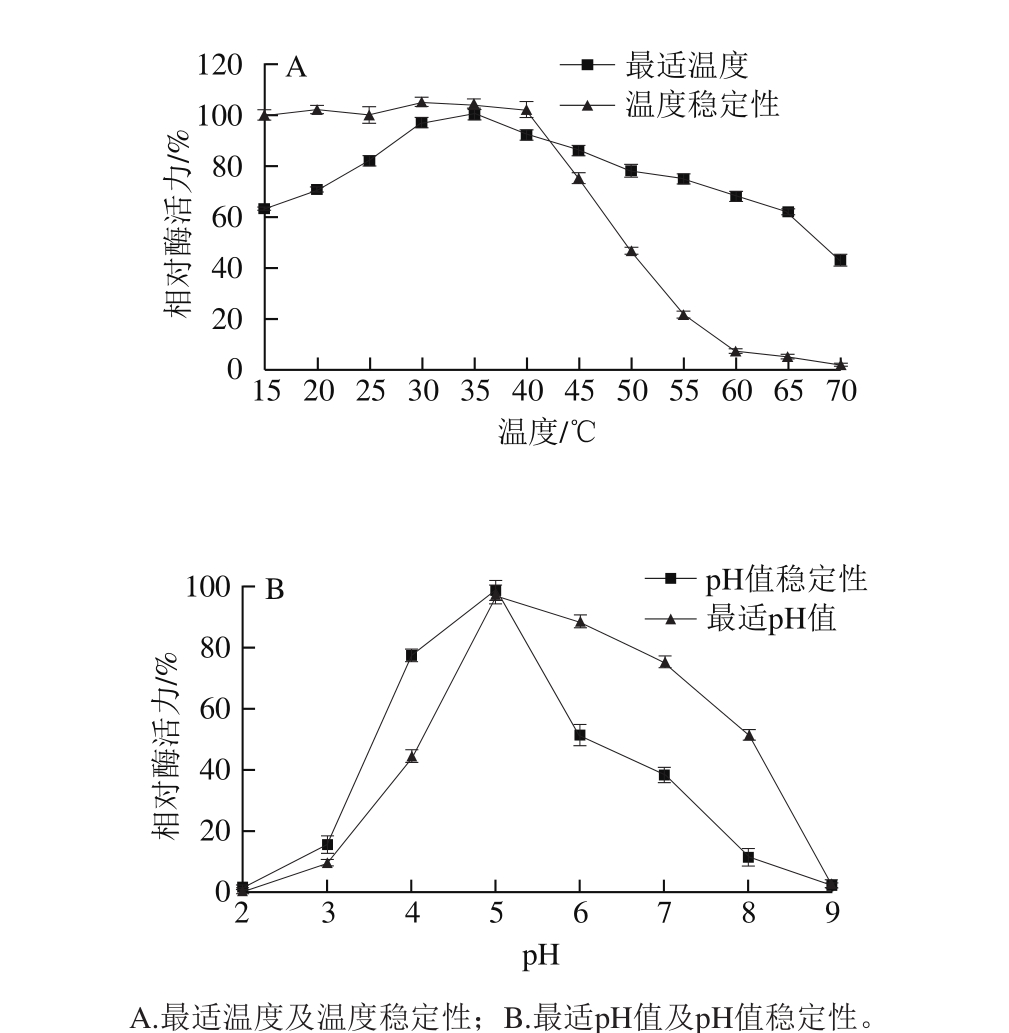
图5 重组右旋糖酐酶酶学性质
Fig. 5 Enzymatic properties of the recombinant dextranase
2.5.2 重组右旋糖酐酶底物特异性
由表3可知,将重组右旋糖酐酶作用于不同的糖类底物,发现了与天然右旋糖酐酶相同的底物特异性,即专一性作用于α-1,6糖苷键,不作用于β-1,4、β-1,2以及α-1,2等其他糖苷键型。以右旋糖酐T70为底物,配制0.1%~1.0%的底物溶液,计算得到重组右旋糖酐酶的K m 为87.56 μmol/L,v max 为428.02 μmol/(min·mg)。
表3 重组右旋糖酐酶对不同糖类底物的作用
Table 3 Action of recombinant dextranase on various substrate carbohydrates

发酵培养环境对酵母分泌表达能力具有很大的影响 [19] ,因此可以尝试通过改善酵母的发酵条件提高酵母分泌表达外源蛋白的能力。首先对重组酵母发酵培养基发酵pH值、诱导剂添加量、表面活性剂含量、碳源添加量、装液量等参数进行单因素优化。结果表明单因素优化后的培养条件使重组酶活力提高了2.6 倍,从初始的89.56 U/mL提高到了240.74 U/mL,总蛋白质量浓度为0.068 mg/mL。
在重组毕赤酵母中甲醇诱导AOX1启动子表达重组右旋糖酐酶,如图6A所示,当甲醇添加量为1.0%(每24 h)时重组酶活力达到122 U/mL。高于1.0%(每24 h)时,随着甲醇添加量的增加,酶活力逐渐降低。通过摇瓶中的装液量控制通气量,实验结果如图6B所示,500 mL锥形瓶中装液量为25 mL、50 mL时产酶量相当,以50 mL时为最佳。发酵pH值优化结果如图6C所示,当pH 5.0时酶活力最高,酸性碱性环境下产酶量急剧降低。低温发酵能够降低中性蛋白酶对目标外源蛋白的降解,促进新生肽的折叠,提高目标蛋白的稳定性,以及减轻发酵过程的氧气压力 [20-21] 。在20、25、30 ℃三个温度梯度下考察右旋糖酐酶活力,如图6D所示,25 ℃时重组右旋糖酐酶活力为30 ℃时的1.6 倍。20 ℃发酵120 h酶活力与25 ℃发酵96 h酶活力相当,以25 ℃为最佳发酵温度。非离子表面活性剂具有增加细胞壁渗透的作用,从而提高重组酵母的分泌能力 [22] 。考察吐温-80、吐温-20两种常见的表面活性剂对酵母分泌的影响,如图6E所示,当吐温-80添加量为4 g/L时,发酵上清液的右旋糖酐酶活力增加到230.74 U/mL。在甲醇发酵诱导阶段,甲醇既作为碳源维持酵母生长,又作为诱导剂诱导外源蛋白的合成 [23] 。在酵母发酵过程中以山梨醇或者甘油为协同底物提供酵母生长所需碳源,以减轻酵母代谢压力 [24-25] 。如图6F所示,以山梨醇作为碳源的效果明显优于甘油,当山梨醇添加量为5 g/L时,重组右旋糖酐酶活力达到240.74 U/mL。
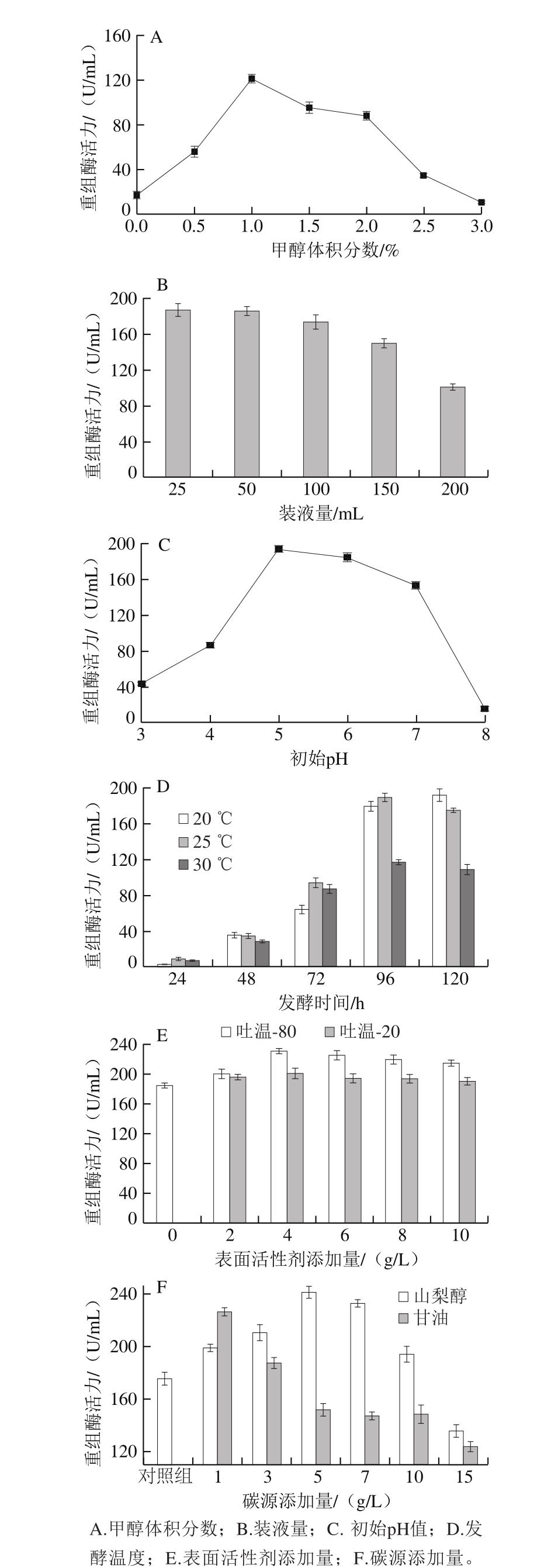
图6 重组毕赤酵母发酵产酶水平的初步摇瓶优化
Fig. 6 Optimization of shake flask culture of the recombinant P. pastoris for dextranase production
目前已报道的右旋糖酐酶编码基因主要来源于青霉菌属、杆状菌属以及链球菌属等,除朱黄青霉外其他青霉来源的右旋糖酐酶基因鲜有报道。因此通过对棘孢青霉来源的右旋糖酐酶的研究可以加深对青霉属右旋糖酐酶的认识。朱黄青霉右旋糖酐酶的最适催化温度为55 ℃,高于天然棘孢青霉酶右旋糖酐酶的35 ℃,但其热稳定性差,在50 ℃条件下保温15 min酶活力损失50%以上 [9] ,而棘孢青霉右旋糖酐酶在50 ℃保温1 h后仍然能保留80%以上的酶活力,并且在最适温度35 ℃条件下保温2 d后仍然能保留88%的酶活力 [16] 。细丽毛壳菌来源的右旋糖酐酶目前已经小规模产业化,该酶最适催化温度为60 ℃、pH 5.5,专一性催化断裂糖酐链中连续的α-1,6糖苷键,但该酶液同样也存在温度稳定性差的现象,65 ℃时半衰期为10 min,60 ℃时半衰期为250 min [26] ,而棘孢青霉右旋糖酐酶的温度稳定性表明在最适催化温度条件下保藏24 h后,仍然能保留93.5%的酶活力,10 d后酶活力降至50%,说明与朱黄青霉右旋糖酐酶以及细丽毛壳菌右旋糖酐酶相比,棘孢青霉右旋糖酐酶虽然具有相对较低的最适催化温度,但其具有更好的温度稳定性,更加适应工业生产长时间的催化要求。
以棘孢青霉菌为模板,反转录合成右旋糖酐酶编码基因,优化序列后在毕赤酵母中进行表达。毕赤酵母分泌表达的重组右旋糖酐酶最适催化温度为35 ℃、最适催化pH值为5.0,具有与天然酶相似的催化性质。但在温度稳定性上略有降低,类似的情况也出现在斯达克油脂酵母(Lipomyces starkeyi 1390)来源的右旋糖酐酶基因的异源表达中,异源表达的重组右旋糖酐酶最适温度为30 ℃,低于天然酶的55 ℃,最适pH 4.5,低于天然酶的5.0 [27-28] ,可能是因为异源表达过程中不同的表达体系对蛋白结构进行了一定程度的修饰,具体原因有待进一步深入研究。
在摇瓶水平上对重组酵母的产酶能力进行单因素优化,优化后重组酶活力达到240.74 U/mL,高于Chen Lin等 [27] 在5 L发酵罐获得83.9 U/mL的重组酶活力,Kang等 [7] 在10 L的发酵罐中获得的134 U/mL的酶活力,低于梁达奉等 [17] 在5 L发酵罐中获得的1 048 U/mL(诱导型)以及398 U/mL(组成型)重组酶活力,但高于其摇瓶单因素优化后的75 U/mL(诱导型)以及60.4 U/mL(组成型)。说明重组棘孢青霉右旋糖酐酶在摇瓶水平的表达能力达到国内现有水平。但是在摇瓶水平总蛋白质量浓度低,仅为0.068 mg/mL,显著低于Roca等 [8] 在10 L发酵罐中表达的右旋糖酐酶产量。但毕赤酵母是一种适合高密度发酵的菌株,在发酵罐中毕赤酵母分泌表达能力能够显著提高,例如里氏木霉Cel5基因在5 L发酵罐中酶活力相比摇瓶提高了11 倍 [29] 、雪白根霉在50 L发酵罐中的酶活力相对于摇瓶发酵提高了40 倍 [30] 。所以可期望通过高密度发酵条件优化,进一步提高重组毕赤酵母分泌表达右旋糖酐酶的能力。
以棘孢青霉基因组为模板克隆得到右旋糖酐酶编码基因,序列优化后整合到毕赤酵母X33中,表达后的右旋糖酐酶具有与天然酶相似的酶学性质,说明重组右旋糖酐酶可以代替天然棘孢青霉右旋糖酐酶直接应用于工业催化制备右旋糖酐。条件优化后重组毕赤酵母摇瓶产酶能力达到240.74 U/mL,达到国内现有摇瓶表达水平。
参考文献:
[1] 张宇琪, 张洪斌, 甘微苇, 等. 右旋糖酐酶研究进展[J]. 生物工程学报, 2015, 31(5): 634-647. DOI:10.13345/j.cjb.140475.
[2] ZHANG H B, GAN W W, ZHANG Y Q, et al. Synthesis of isomaltooligosaccharides by using recombinant dextransucrase and Hypocrea lixii dextranase[J]. Journal of Chemical & Pharmaceutical Research,2013, 5(11): 49-53.
[3] KIM Y M, SEO M Y, KANG H K, et al. Construction of a fusion enzyme of dextransucrase and dextranase: application for one-step synthesis of isomalto-oligosaccharides[J]. Enzyme &Microbial Technology, 2009, 44(3): 159-164. DOI:10.1016/j.enzmictec.2008.10.007.
[4] THITARAM S N, CHUNG C H, DAY D F, et al.Isomaltooligosaccharide increases cecal bifidobacterium population in young broiler chickens[J]. Poultry Science, 2005, 84(7): 998.DOI:10.1093/ps/84.7.988.
[5] WANG X, CHENG H, LU M, et al. Dextranase from Arthrobacter oxydans KQ11-1 inhibits biofilm formation by polysaccharide hydrolysis[J]. Biofouling, 2016, 32(10): 1223-1233. DOI:10.1080/089 27014.2016.1239722.
[6] OTSUKA R, IMAI S, MURATA T, et al. Application of chimeric glucanase comprising mutanase and dextranase for prevention of dental biofilm formation[J]. Microbiology & Immunology, 2015,59(1): 28-36. DOI:10.1111/1348-0421.12214.
[7] KANG H K, PARK J Y, AHN J S, et al. Cloning of a gene encoding dextranase from Lipomyces starkeyi and its expression in Pichia pastoris[J]. Journal of Microbiology & Biotechnology, 2009, 19(2):172-177. DOI:10.4014/jmb.0802.100.
[8] ROCA H, GARCIA B, RODRIGUEZ E, et al. Cloning of the Penicillium minioluteum gene encoding dextranase and its expression in Pichia pastoris[J]. Yeast, 1996, 12(12): 1187-1200.DOI:10.1002/(SICI)1097-0061(19960930)12:12<1187::AIDYEA986>3.0.CO;2-U.
[9] 梁达奉. α-葡聚糖酶的基因工程菌构建、发酵及其应用研究[D].广州: 广东工业大学, 2011: 20-50. DOI:10.7666/d.y2028007.
[10] 黄曾慰, 梁达奉, 曾练强, 等. 朱黄青霉α-葡聚糖酶在毕赤酵母中的高效表达[J]. 广西科学, 2014(6): 614-618. DOI:10.13656/j.cnki.gxkx.2014.06.006.
[11] ABDEL-NABY M A, ISMAIL A M S, ABDEL-FATTAH A M,et al. Preparation and some properties of immobilized Penicillium funiculosum 258 dextranase[J]. Process Biochemistry, 1999, 34(4):391-398. DOI:10.1016/S0032-9592(98)00127-7.
[12] SZCZODRAK J, PLESZCZYNSKA M, FIEDUREK J. Penicillium notatum 1 a new source of dextranase[J]. Journal of Industrial Microbiology and Biotechnology, 1994, 13(5): 315-320.
[13] PLESZCZYNSKA M, ROGALSKI J, SZCZODRAK J, et al.Purification and some properties of an extracellular dextranase from Penicillium notatum[J]. Mycological Research, 1996, 100(6): 681-686.DOI:10.1016/S0953-7562(96)80198-5.
[14] ZHANG Y Q, LI R H, ZHANG H B, et al. Purification,characterization, and application of a thermostable dextranase from Talaromyces pinophilus[J]. Journal of Industrial Microbiology &Biotechnology, 2016, 44(2): 1-11. DOI:10.1007/s10295-016-1886-8.
[15] ERHARDT F A, STAMMEN S, JÖRDENING H J. Production,characterization and (co-)immobilization of dextranase from Penicillium aculeatum[J]. Biotechnology Letters, 2008, 30(6): 1069-1073. DOI:10.1007/s10529-008-9659-8.
[16] 张洪斌, 吴定涛, 黄丽君, 等. 一株产右旋糖酐酶青霉的分离及酶的纯化和性质[J]. 微生物学报, 2011, 51(4): 495-503. DOI:10.13343/j.cnki.wsxb.2011.04.006.
[17] 王建荣, 刘丹妮, 夏雨, 等. 密码子优化及透明颤菌血红蛋白共表达提高耐热脂肪酶在毕赤酵母的表达[J]. 食品科学, 2016, 37(19):135-140. DOI:10.7506/spkx1002-6630-201619023.
[18] BRADFORD M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of proteindye binding[J]. Analytical Biochemistry, 1976, 72: 248-254.
[19] BEN AZOUN S, KALLEL H. Investigating the effect of carbon source on rabies virus glycoprotein production in Pichia pastoris by a transcriptomic approach[J]. Microbiologyopen, 2017, 6(4): 1-9.DOI:10.1002/mbo3.489.
[20] YANG Z, ZHANG Z. Engineering strategies for enhanced production of protein and bio-products in Pichia pastoris: a review[J]. Biotechnol Advance, 2018, 36(1): 182-195. DOI:10.1016/j.biotechadv.2017.11.002.
[21] 槐强强, 贾禄强, 丁健, 等. 通过在低细胞浓度下启动甲醇诱导、优化碳/能量代谢模式促进毕赤酵母表达Monellin[J]. 生物工程学报,2018, 34(2): 282-293. DOI:10.13345/j.cjb.170189.
[22] LIU Y, XUAN S, LONG C, et al. Screening, identifying of cellulosedecomposing strain L-06 and its enzyme-producing conditions[J].Chinese Journal of Biotechnology, 2008, 24(6): 1112-1116.DOI:10.13345/j.cjb.2008.06.017.
[23] LOOSER V, BRÜHLMANN B, BUMBAK F, et al. Cultivation strategies to enhance productivity of Pichia pastoris: a review[J].Biotechnology Advances, 2015, 33(6): 1177-1193. DOI:10.1016/j.biotechadv.2015.05.008.
[24] 汪汇慧, 金虎, 高敏杰, 等. 甲醇/山梨醇共混流加诱导改变毕赤酵母生产猪α-干扰素过程的代谢产能途径强化发酵性能[J]. 生物工程学报, 2012, 28(2): 164-177. DOI:10.13345/j.cjb.2012.02.003.
[25] 王丙莲, 冯东, 梁晓辉, 等. 甘油及甲醇补料策略对毕赤酵母表达猪α-干扰素的影响[J]. 激光生物学报, 2016, 25(6): 553-558.DOI:10.3969/j.issn.1007-7146.2016.06.011.
[26] 麻少莹. 细丽毛壳菌发酵生产右旋糖酐酶的工艺条件优化及应用研究[D]. 南宁: 广西大学, 2014: 20-50. DOI:10.7666/d.D524754.
[27] CHEN L, ZHOU X S, FAN W M, et al. Expression, purification and characterization of a recombinant Lipomyces starkey dextranase in Pichia pastoris[J]. Protein Expression and Purification, 2008, 58(1):87-93. DOI:10.1016/j.pep.2007.10.021.
[28] KOENIG D W, DAY D F. Induction of Lipomyces starkeyi dextranase[J]. Applied & Environmental Microbiology, 1989,55(8): 2079.
[29] 白仁惠, 张云博, 王春迪, 等. 里氏木霉Cel5A基因优化及其在毕赤酵母中的高效表达[J]. 生物工程学报, 2016, 32(10): 1381-1394.DOI:10.13345/j.cjb.160017.
[30] 王建荣, 刘丹妮, 夏雨, 等. 优化密码子及诱导温度提高雪白根霉脂肪酶在毕赤酵母中的表达[J]. 食品与发酵工业, 2017, 43(1): 18-23.DOI:10.13995/j.cnki.11-1802/ts.201701004.
Cloning of the Penicillium aculeatum Gene Encoding Dextranase and Its Expression in Pichia pastoris
WU Min, ZHANG Yuxin, HU Xueqin, ZHANG Hongbin*
(School of Biological and Medical Engineering, Hefei University of Technology, Hefei 230009, China)
Abstract: The dextranase-encoding gene (dex) was amplified by reverse transcription PCR from the genome of Penicillium aculeatum F1001. The gene consisted of 1 866 base pairs and encoded a protein of 622 amino acid residues. According to the codon usage bias of Pichia pastoris, optimized gene (opt-dex) was obtained. The expression recombinant plasmids dexpPICZαA and opt-dex-pPICZαA were constructed and then separately electro-transformed into P. pastoris X33 to form transformants. The dextranase-producing transformants were selected out using blue-dextran T-2000 specific plates and shake flask expression. The purified recombinant dextranase showed only one band about 65 kDa, as analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The recombinant dextranase reacted optimally at pH 5 and 35 ℃ and specifically cleaved α-1,6 glycosidic bonds. The shake-flask fermentation conditions of the recombinant strain were optimized as follows: temperature 25 ℃, initial pH 5.0, addition of 1% (V/V) methanol, 4 g/L Tween-80 and 5 g/L sorbitol at 24 h intervals, and 50 mL of medium contained in a 500-mL shake flask. Under the optimized conditions, the activity of dextranase was as high as 240.74 U/mL. In conclusion, this study indicates that P. pastoris X33 is suitable for heterologous expression of P. aculeatum dextranase and that the recombinant dextranase can be used as an alternative to the native dextranase in producing dextran for industrial application.
Keywords: dextranase; optimization; Pichia pastoris; expression
WU Min, ZHANG Yuxin, HU Xueqin, et al. Cloning of the Penicillium aculeatum gene encoding dextranase and its expression in Pichia pastoris[J]. Food Science, 2018, 39(18): 73-80. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201818012. http://www.spkx.net.cn
吴敏, 张宇馨, 胡雪芹, 等. 棘孢青霉右旋糖酐酶基因克隆及其在毕赤酵母中的表达[J]. 食品科学, 2018, 39(18): 73-80.DOI:10.7506/spkx1002-6630-201818012. http://www.spkx.net.cn
文章编号: 1002-6630(2018)18-0073-08
引文格式:
中图分类号: Q814
文献标志码: A
*通信作者简介: 张洪斌(1970—),男,教授,博士,研究方向为生物制药与酶工程。E-mail:zhb5678@163.com
DOI: 10.7506/spkx1002-6630-201818012
基金项目: 国家自然科学基金面上项目(81573399)
第一作者简介: 吴敏(1993—),女,硕士研究生,研究方向为生物制药与酶工程。E-mail:1545218573@qq.com
收稿日期: 2018-01-17