
图1 gud1和egud编码区的扩增
Fig. 1 Amplification of encoding regions of gud1 and egud genes
孙 莹,潘思安,李萌萌,邓威威,张正竹*
(安徽农业大学 茶树生物学与资源利用国家重点实验室,安徽 合肥 230036)
摘 要: 与植物体内合成路径不同,微生物体内合成咖啡碱存在一条以黄嘌呤为底物,利用鸟嘌呤脱氨酶催化鸟嘌呤生成黄嘌呤有效合成咖啡碱的新途径。为克隆鸟嘌呤脱氨酶的基因,构建可高效合成黄嘌呤的原核表达载体并对外源蛋白活性进行检测,分别以酿酒酵母和大肠杆菌为研究材料,根据GenBank中酿酒酵母和大肠杆菌中鸟嘌呤脱氨酶基因gud1和egud序列设计引物,聚合酶链式反应特异扩增其基因片段,将目的基因连接至pMAL-c5X载体,转入大肠杆菌BL21(DE3)中诱导蛋白表达,并用高效液相色谱法鉴定其目的蛋白的催化活性。结果表明重组载体pMAL-gud1、pMAL-egud均可用来合成黄嘌呤,且GUD1比EGUD合成黄嘌呤的效率更高。研究结果将进一步丰富黑茶加工技术理论,同时为体外构建高效咖啡碱生物工程菌提供理论支持。
关键词: 黑茶加工;咖啡碱;鸟嘌呤脱氨酶;黄嘌呤;生物合成;原核表达
咖啡碱是茶叶中含量最高的生物碱,也是茶叶重要的滋味物质 [1] ,能兴奋中枢神经从而消除疲劳感,还具有抑菌、抗癌、抗肥胖等功效 [2] 。在绿茶及乌龙茶这些不发酵茶或为发酵茶的加工过程中,咖啡碱含量基本保持稳定或有些微的降低 [3-4] ,而在普洱茶和茯茶等黑茶加工中,由于大量微生物参与了渥堆发酵过程,导致产品中咖啡碱含量的明显增长 [5-6] 。咖啡碱含量的消长是黑茶品质形成及其健康功能的重要物质基础。目前,国际市场上的咖啡碱主要来源于化学合成,受潜在的化学污染物质量安全风险影响,其在食品行业和医药产业中的应用受到限制 [7] 。因此,污染低、安全性高的生物合成咖啡碱越来越受关注。
在富含咖啡碱的植物,如咖啡和茶树中,咖啡碱合成路径是黄嘌呤核苷→7-甲基黄嘌呤核苷→7-碱基黄嘌呤→可可碱→咖啡碱,即是以黄嘌呤核苷为底物,以N-7、N-3、N-1顺序进行3 次甲基化 [8-10] 。此外也有研究指出微生物中咖啡碱合成可能还存在一条有别于植物体内合成咖啡碱的途径,以N-3、N-1、N-7的顺序进行甲基化,以黄嘌呤为底物逐步生成咖啡碱 [11-12] 。因此,在微生物中增加咖啡碱合成途径中黄嘌呤的含量,对实现咖啡碱无底物从头合成及提高咖啡碱产量都具有重要的现实意义。
鸟嘌呤脱氨酶又称鸟嘌呤酶或鸟嘌呤氨基水解酶,是一种锌金属酶 [13] ,也是核苷酸代谢的关键酶,分别在1982、1998年及1999年被证实可在脉孢菌、嗜盐古菌和哺乳类动物中催化鸟嘌呤脱氨降解生成黄嘌呤后合成核苷酸 [14-17] 。嘌呤、嘧啶在作为碳源及氮源时,脱氨步骤是第一步,这也是降解途径和补偿途径的基础步骤 [18] 。但是因为嘌呤代谢的补偿途径是不对称的,只有腺嘌呤衍生物可以转变成鸟嘌呤核苷酸,而相反的路径却不存在,因此鸟嘌呤脱氨酶在鸟嘌呤分解代谢中扮演着重要角色 [19-20] 。植物体中关于鸟嘌呤脱氨酶的研究较少,Negishi等 [21] 曾在茶树叶片中检测到了鸟嘌呤脱氨酶活性,只是尚未发现完整的基因序列。目前关于鸟嘌呤脱氨酶的研究主要集中在动物肝脏及微生物方面 [22-23] 。研究显示,虽然鸟嘌呤脱氨酶是嘌呤代谢基础酶,但是在哺乳动物体内分布并不均匀,例如在小鼠体内是在小肠近端部分含有最高水平,仅在小鼠脑内不同区域鸟嘌呤脱氨酶含量就有近50 倍的差异 [16,24-26] 。本研究分别从酿酒酵母和大肠杆菌中克隆出鸟嘌呤脱氨酶基因gud1和egud,构建重组载体进行原核表达分析,尝试在生物体内大量积累黄嘌呤,通过高效液相色谱(high performance liquid chromatography,HPLC)法检测体内及体外产物,分析鉴定基因功能,尝试为丰富黑茶加工技术理论作出探索,同时为无底物合成咖啡碱和提高咖啡碱产量提供理论依据。
表达菌株大肠杆菌BL21(DE3)、感受态大肠杆菌TransT1、DNA Marker、Protein Marker II 北京全式金公司;表达载体pMAL-c5X由安徽农业大学茶树生物学与资源利用国家重点实验室提供;rTaq酶、限制性内切酶 宝生物工程(大连)有限公司;ClonExpress ® II One Step Cloning Kit 诺唯赞生物科技有限公司;引物合成及测序由通用生物系统(安徽)有限公司完成;HPLC所用试剂均为色谱级,其他试剂均为国产分析纯。
Alliance高效液相色谱仪、ODS C 18 反相柱(250 mm×4.6 mm,5 μm) 美国Waters公司;S1000 TM Thermal Cycler聚合酶链式反应(polymerase chain reaction,PCR)仪、凝胶成像系统(ChemiDoc TM MP Imaging System)、Power PAC 3000电泳仪 美国Bio-Rad公司;JY92-IIN超声波细胞破碎仪 宁波新芝仪器研究所;5424R高速冷冻离心机 德国Eppendorf公司;DHG-9031A电热恒温培养箱 上海精宏实验设备有限公司;LDZF-50L高压灭菌锅 上海申安医疗器械厂。
1.3.1 利用ClonExpress ® II One Step Cloning Kit克隆目的基因
根据gud1和egud序列,按试剂盒说明,利用CE Design V1.04软件设计两对特异性引物,将gud1和egud分别连接到pMAL-C5X。为方便连接目的基因和表达载体,在引物中引入BamHI酶切位点,引物序列如下(下划线处为添加的酶切位点):egud-F:5’-cgcgatatc gtcgacggatccATGATGTCAGGAGAACACACGTTA-3’,egud-F:5’-acctgcagggaattcggatccTTAGTTGCGTTCGTA CACCAGACG-3’;gud1-F:5’-cgcgatatcgtcgacggatccATG ACAAAAAGTGATTTATTATTTGATA-3’,gud1-F:5’-ac ctgcagggaattcggatccCTAAATCTGGTAGACTTGCTGG CC-3’。
以大肠杆菌BL21(DE3)菌株和酿酒酵母INVSCI菌株为试材,因两种菌株皆没有内含子,直接以粗样品为模板,用KOD FX Neo扩增egud和gud1。PCR体系:2×PCR buffer 25 µL,2 mmol/L dNTPs 10 µL,KOD FX Neo 1 µL, Primer F(10 µmol/L)1.5 µL ,Primer R(10 µmol/L)1.5 µL,补ddH 2 O至50 µL,超净台中挑菌少许,该体系均分成2 管进行PCR。PCR程序(35 个循环)如下:94 ℃ 2 min,98 ℃ 10 s,58 ℃ 30 s,68 ℃ 80 s,68 ℃ 10 min,4 ℃ ∞。PCR产物用1.2%琼脂糖凝胶电泳检测,回收目的条带。
1.3.2 原核表达载体的构建与鉴定
对表达载体质粒pMAL-c5X进行单酶切,酶切体系为:10×K buffer 2 µL,BamHI 1 µL,DNA 2~5 µL(不大于1 µg),补ddH 2 O至20 µL。上述体系30 ℃水浴条件下反应1 h,产物经电泳检测后,用胶回收试剂盒纯化产物。与PCR回收产物重组连接,反应体系:5×CE II buffer 4 µL,Exnase ® II 2 µL,Linearized Vector 5 µL,Purified PCR Fragment 0.5 µL,补ddH 2 O至20 µL。上述体系于37 ℃反应30 min进行连接,反应完成后及时冰浴5 min终止反应,将10 µL连接产物转入50 µL大肠杆菌TransT1感受态细胞,冰浴30 min,然后将离心管置于45 ℃水浴45 s,冰浴中冷却2 min。在超净工作台中分别向离心管中加入500 µL LB培养基,混匀后37 ℃条件下200 r/min培养1 h。1 h后吸取100 µL菌液均匀涂布到含有氨苄青霉素的LB平板上,37 ℃过夜培养。挑取单菌落于含氨苄抗性的LB固体培养基平板上划线,37 ℃培养后蘸取少量菌体进行菌落PCR,挑取阳性菌落送通用生物系统(安徽)有限公司测序。将测序正确的阳性菌落挑菌初步培养,用试剂盒提取重组质粒转入至大肠杆菌BL21(DE3)。
1.3.3 重组蛋白的诱导表达
筛选阳性菌落于3 mL LB培养基中,37 ℃条件下200 r/min振荡过夜培养。次日取800 µL过夜菌液接种于40 mL新鲜LB培养基,继续37 ℃振荡培养至OD值为0.6~0.8。取1 mL菌液于12 000 r/min下离心5 min后,取上清液作为诱前对照。在剩余菌液中加入终浓度为1 mmol/L的异丙基硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)进行诱导,分别16 ℃诱导20 h,30 ℃诱导8 h,确定最佳诱导温度及时间。
1.3.4 酶活性检测
1.3.4.1 体外酶活性检测
选取最佳诱导条件,进行融合蛋白的诱导表达,200 mL诱导后菌液冷冻离心(4 ℃,6 000 r/min)20 min收集菌体。加入20 mL Lysis Buffer(现加二硫苏糖醇,终浓度为5 mmol/L)重悬菌体,冰浴条件进行超声破碎,破碎结束离心得上清液即粗酶液,将粗酶液过麦芽糖结合MBP层析柱进行纯化。
5 mL酶反应体系 [20] :MgCl 2 250 μL,鸟嘌呤溶液50 μL,纯化后的酶液200 μL,无菌水4.5 mL,混合体系37 ℃反应,分别取反应0、5、10、30、60 min的样品,离心后过0.22 µm水相滤膜,通过HPLC检测酶反应产物。以未插入目的基因的空载体pMAL-c5X相同处理作对照。
1.3.4.2 体内酶活性检测
40 mL菌液诱导后,分为两组,一组不作处理,一组加入4 mL质量浓度为5 mg/mL的鸟嘌呤为底物,将菌液移入37 ℃继续培养,每隔24 h取一次样,将菌液12 000 r/min离心10 min,按1∶50的比例取上清液稀释,稀释液过0.22 µm水相滤膜,通过HPLC检测酶反应产物。
高效液相色谱条件:流速1 mL/min;流动相:A相0.2%乙酸,B相纯乙腈;紫外检测器吸收波长为274 nm。线性变化范围:0~4 min,A相95%,B相5%;4~10 min,A相40%,B相60%;10~13 min,A相95%,B相5%;13~30 min,A相95%,B相5%。
1.3.5 表达酶的同源建模以及二级结构预测
采用DNAMAN软件进行GUD1和EGUD的氨基酸序列进行分析比对比,参考Dai Xinlong等 [27] 的建模方法,使用SWISS-MODEL在线软件(http://swissmodel.expasy.org/interactive)利用相似度高的已知三维结构的蛋白酶的晶体结构模型,对GUD1和EGUD进行蛋白三维结构的同源建模,并使用Molegro virtual docker软件以此对目的蛋白二级结构进行了预测。
PCR扩增图像、结合蛋白诱导表达图像及蛋白纯化图像都是利用伯乐ChemiDoc™MP Imaging System照胶成像所得并进行后续编辑;色谱图为HPLC仪检测编辑并用Excel进行后续处理;蛋白晶体模型使用Molegro virtual docker软件进行编辑。
挑取少量大肠杆菌BL21(DE3)和酿酒酵母INVSCI,用KOD FX Neo分别扩增gud1和egud(图1),在1 000~2 000 bp之间分别可见两条特异性条带,产物大小与gud1 ORF的长度1 470 bp、egud ORF的长度1 320 bp相符。回收PCR扩增产物,与pMAL-c5X表达载体通过Exnase ® II重组连接,构建pMAL-gud1和pMAL-egud表达载体。
图1 gud1和egud编码区的扩增
Fig. 1 Amplification of encoding regions of gud1 and egud genes
为确定EGUD和GUD1蛋白最佳诱导条件,加入1 mmol/L IPTG在16 ℃和30 ℃分别诱导20 h和8 h,制样进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDSPAGE)检测。在两个温度下均可诱导得到90 kDa左右的可溶性目的蛋白(图2),与融合蛋白大小一致(空载标签子MBP大小为40 kDa,EGUD和GUD1蛋白理论值大小51.22 kDa和55.21 kDa)。而且16 ℃诱导20 h蛋白表达效果明显比30 ℃诱导效果好,融合蛋白大量表达在上清液中,因此后续实验均在16 ℃、20 h诱导条件下进行。将菌液诱导结束后制取粗酶液,利用麦芽糖结合MBP层析柱进行纯化,获得单一条带(图3)。

图2 pMAL-egud和 pMAL-gud1诱导蛋白SDS-PAGE图
Fig. 2 Induced protein expression of pMAL-egud and pMAL-gud1 revealed by SDS-PAGE electrophoresis
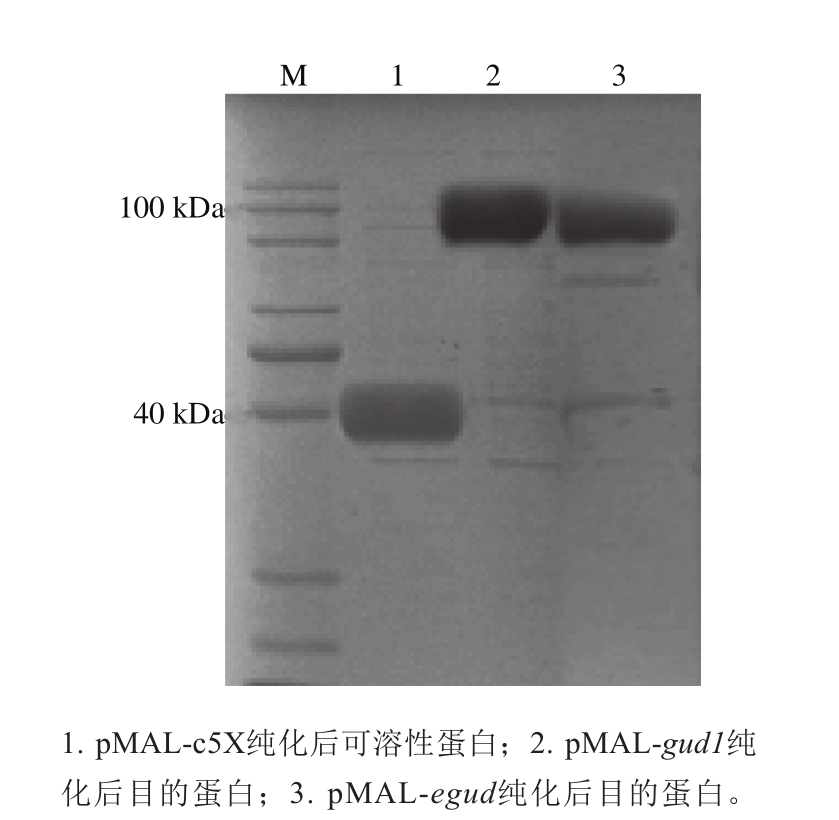
图3 pMAL-gud1、pMAL-egud目的蛋白纯化
Fig. 3 Purification of target protein pMAL-gud1 and pMAL-egud


图4 pMAL-gud1和pMAL-egud产物体外酶活性的HPLC分析
Fig. 4 In vitro activity analysis of pMAL-gud1 and pMAL-egud by HPLC
如图4所示,对照组pMAL-c5X无法生成黄嘌呤,与对照组相比,样品组pMAL-gud1和pMAL-egud添加的底物鸟嘌呤均有所下降并且生成黄嘌呤质量浓度分别达到了619.3 μg/mL和527.9 μg/mL。

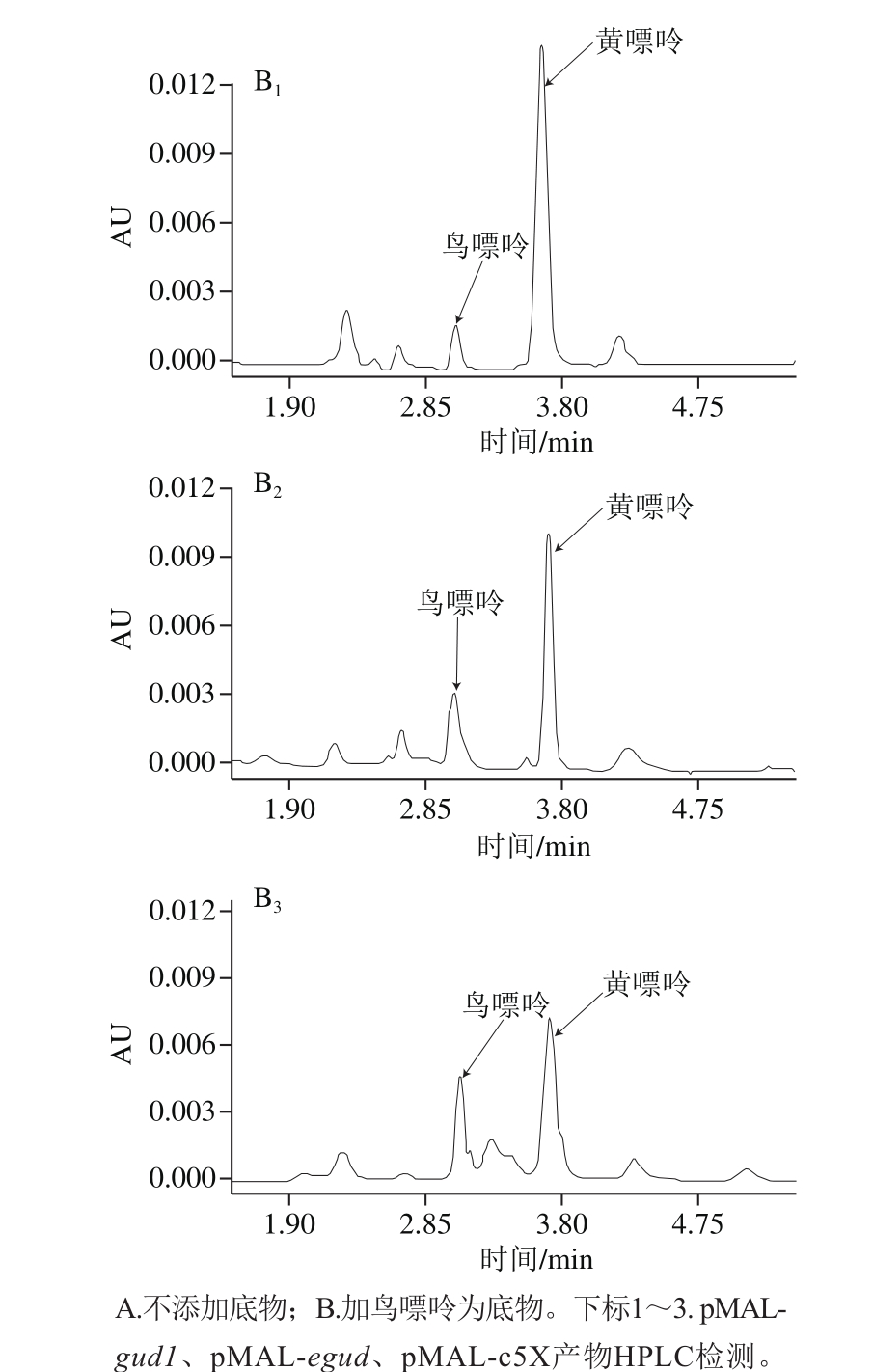
图5 pMAL-gud1和pMAL-egud产物体内酶活性的HPLC分析
Fig. 5 In vivo activity of pMAL-gud1 and pMAL-egud analyzed by HPLC
菌液诱导后制样通过HPLC检测EGUD和GUD1体内酶活性,结果见图5。在两种处理下,对照组中转入空载的菌株也都能产生黄嘌呤,但是与重组菌株相比较发现,无底物培养时重组菌株产物明显黄嘌呤产量高于对照组,转入pMAL-gud1和pMAL-egud质粒的实验组菌株对照转入空载质粒的对照组菌株,黄嘌呤产量由110 μg/mL分别提高到225 μg/mL和191 μg/mL;加底物培养时实验组底物鸟嘌呤消耗速度相比对照组更快,且黄嘌呤的产量更高,转入pMAL-gud1和pMAL-egud质粒的实验组菌株对照转入空载质粒的对照组菌株,黄嘌呤产量由180 μg/mL分别提高到312 μg/mL和267 μg/mL。
通过SWISS-MODEL在线软件对GUD1和EGUD蛋白进行同源建模,分别以Guad(SMTL id:2uz9.1)和Blr3880(SMTL id:200D.1)蛋白为模型,建造了GUD1和EGUD的三维晶体模型(图6),重合度分别达到了41.15%和44.72%,并以此对二者的二级结构进行了预测,如图7所示,GUD1二级结构以α-螺旋为主,α-螺旋较多较集中,EGUD二级结构中α-螺旋与β-折叠程度相似,但是β-折叠较α-螺旋更为分散。

图6 GUD1和EGUD三维晶体同源建模
Fig. 6 3D crystal modeling of GUD1 and EGUD

图7 GUD1和EGUD二级结构预测
Fig. 7 Predicted secondary structures of GUD1 and EGUD
本研究在大肠杆菌中分别转入酿酒酵母的鸟嘌呤脱氨酶基因gud1和大肠杆菌鸟嘌呤脱氨酶基因egud构建重组质粒。由于低温有利于增强蛋白的稳定性和正确折叠,获得可溶性蛋白的可能性较大,而不易形成包涵体 [28] ,所以实验过程中利用IPTG在16 ℃低温诱导,从而使大肠杆菌中可以大量产生黄嘌呤。本研究构建的工程菌在转入含gud1和egud的质粒后,对比转入空载pMAL-c5X的大肠杆菌,在加入鸟嘌呤作底物时,120 h后黄嘌呤产量由180 μg/mL分别提高到312 μg/mL和267 μg/mL,鸟嘌呤消耗速度分别提高了61%和49%;在不加入底物的情况下,120 h后黄嘌呤产量由110 μg/mL分别提高到225 μg/mL和191 μg/mL。对分别转入gud1和egud的菌株相进行比较,前者生成黄嘌呤的效率更高,说明GUD1比EGUD催化效率更高,有更高的酶活性。可能是因为鸟嘌呤脱氨酶催化鸟嘌呤脱氨生成黄嘌呤是初级代谢反应,相比于大肠杆菌,酵母是真核生物,有更为复杂的生理代谢,导致基础代谢也更为旺盛,或鸟嘌呤脱氨酶有不同的折叠方式和晶体结构,在真核生物和原核生物中因三维结构不同造成GUD1比EGUD酶活性更高 [29] 。后续研究中可以考虑通过以下措施优化基因表达,提高黄嘌呤表达量:控制培养条件以提高质粒稳定性与拷贝数,减少代谢副产物积累,提高外源蛋白产率;更换表达载体,将Lac启动子换成更强的T7启动子等 [30-31] 。
与绿茶等不发酵或轻微发酵茶相比,茶叶中内源酶在杀青过程中已经失活,渥堆和发花过程中,微生物来源的外源酶成为发酵中最主要的生物催化剂,在适宜的水分和温度条件下,黑曲霉、冠突散囊菌、酵母等微生物以茶叶中存留的鸟嘌呤等碱基为底物合成了黄嘌呤,为渥堆和发花过程中咖啡碱的生物合成提供了底物 [32] 。
目前,咖啡碱在植物中的生物合成途径已经基本明确,但是构建工程菌生物合成咖啡碱应还有待进一步研究。本研究以黄嘌呤为底物合成咖啡碱的新途径入手,通过提高咖啡碱合成途径中底物黄嘌呤的生成量,以期达到提高咖啡碱合成量的目标。研究结果表明,无论是否添加外源底物(鸟嘌呤),转入gud1和egud的菌株都明显比对照菌株生成了更多的黄嘌呤。研究结果将为黑茶渥堆技术提供一定的理论基础,同时为体外构建高效咖啡碱生物工程菌提供依据。
参考文献:
[1] 宛晓春. 茶叶生物化学[M]. 北京: 中国农业出版社, 2003.
[2] 程启坤. 茶叶内含成分与人体健康[J]. 世界农业, 2015(5): 199-202.
[3] 许四五, 江平. 绿茶加工技术与主要化学成分的协调研究[J].安徽农业科学, 2012, 40(29): 14459-14460. DOI:10.3969/j.issn.0517-6611.2012.29.105.
[4] 罗学平, 李丽霞, 赵先明, 等. 不同焙火处理对四川乌龙茶香味与化学品质的影响[J]. 食品科学, 2016, 37(17): 104-108. DOI:10.7506/spkx1002-6630-201617017.
[5] WANG X, HU S, WAN X, et al. Effect of microbial fermentation on caffeine content of tea leaves[J]. Journal of Agricultural & Food Chemistry, 2005, 53(18): 7238-7242. DOI:10.1021/jf050495h.
[6] YAN M A, ZHANG D, JIAHUA L I, et al. Determination and comparison of caffeine mass fraction in post-fermented Pu-erh tea,black tea and green tea[J]. Journal of Yunnan Agricultural University,2013, 28(5): 745-749. DOI:10.3969/j.issn.1004-390X(n).2013.05.023.
[7] 武鑫. 茶树咖啡碱生物合成关键酶基因的原核表达及功能验证[D].合肥: 安徽农业大学, 2016.
[8] MCKEAGUE M, WANG Y H, CRAVENS A, et al. Engineering a microbial platform for de novo biosynthesis of diverse methylxanthines[J]. Metabolic Engineering, 2016, 38: 191-203.DOI:10.1016/j.ymben.2016.08.003.
[9] KATO M, KANEHARA T, et al. Caffeine biosynthesis in young leaves of Camellia sinensis: in vitro studies on N-methyltransferase activity involved in the conversion of xanthosine to caffeine[J]. Physiologia Plantarom, 1996, 98: 629-636. DOI:10.1111/j.1399-3054.1996.tb05720.x.
[10] SUMKI T, ASHIHARA H, WALLER G R. Purine and purine alkaloid metabolism in Camellia and Coffea plants[J]. Phytochemistry, 1992,3l(8): 2575-2584. DOI:10.1016/0031-9422(92)83590-U.
[11] WANG X G, WAN X C, HU S X, et al. Study on the increase mechanism of the caffeine content during the fermentation of tea with microorganisms[J]. Food Chemistry, 2008, 107(3): 1086-1091.DOI:10.1016/j.foodchem.2007.09.023.
[12] 谢果, 何蓉蓉, 栗原博. 茶叶生物碱的生物合成与代谢的研究进展[J]. 中国天然药物, 2010, 8(2): 153-160. DOI:10.3724/SP.J.1009.2010.00153.
[13] YAO L S, CUKIER R I, YAN H G. Catalytic mechanism of guanine deaminase: an ONIOM and molecular dynamics study[J]. Journal of Physical Chemistry B, 2007, 111(16): 4200-4210. DOI:10.1021/jp0673056.
[14] MAGILL C W, SABINA R L, GARBER T L, et al. Guanine uptake and metabolism in Neurospora crassa[J]. Journal of Bacteriology,1982, 149(3): 941-947.
[15] STUER-LAURIDSEN B, NYGAARD P. Purine salvage in two halophilic archaea: characterization, of salvage pathways and isolation of mutants,resistant to purine analogs[J]. Journal of Bacteriology, 1998, 180(3): 457.
[16] YUAN G, BIN J C, MCKAY D J, et al. Cloning and characterization of human guanine deaminase[J]. Journal of Biological Chemistry,1999, 274(12): 8175-8180. DOI:10.1074/jbc.274.12.8175.
[17] NYGAARD P, BESTED S M, ANDERSEN K A, et al. Bacillus subtilis guanine deaminase is encoded by the yknA gene and is induced during growth with purines as the nitrogen source[J]. Microbiology,2000, 146(4): 3061-3069. DOI:10.1099/00221287-146-12-3061.
[18] LIAW S H, CHANG Y J, LAI C T, et al. Crystal structure of Bacillus subtilis guanine deaminase: the first domain-swapped structure in the cytidine deaminase superfamily[J]. Journal of Biological Chemistry,2004, 279(34): 35479. DOI:10.1074/jbc.M405304200.
[19] ASHIHARA H, CROZIER A. Biosynthesis and metabolism of caffeine and related purine alkaloids in plants[J]. Advances in Botanical Research,1999, 30(8): 117-205. DOI:10.1016/S0065-2296(08)60228-1.
[20] SAINT-MARC C, DAIGNAN-FOMIER B. GUD1, (YDL238c)encodes Saccharomyces cerevisiae, guanine deaminase, an enzyme expressed during post-diauxic growth[J]. Yeast, 2004, 21(16): 1359-1363. DOI:10.1002/yea.1186.
[21] NEGISHI O, OZAWA T, IMAGAWA H. Guanosine deaminase and guanine deaminase from tea [Camellia sinensis] leaves[J]. Bioscience Biotechnology &Biochemistry, 1994, 58(7): 1277-1281. DOI:10.1271/bbb.58.1277.
[22] 张一兵, 张淑芹, 周文兴, 等. 鸟嘌呤脱氨酶速率法测定及其对肝病的诊断价值[J]. 检验医学, 2010, 25(8): 625-627. DOI:10.3969/j.issn.1673-8640.2010.08.014.
[23] SEFFERMICK J L, DDOGE A G, SADOWSKY M J, et al.Bacterial ammeline metabolism via guanine deaminase[J]. Journal of Bacteriology, 2010, 192(4): 1106-1112. DOI:10.1128/JB.01243-09.
[24] MOHAMEDALI K A, GUICHERIT O M, KELLEMS R E, et al. The highest levels of purine catabolic enzymes in mice are present in the proximal small intestine[J]. Journal of Biological Chemistry, 1993, 268(31): 23728.
[25] BERGER S J, CARTER J G, LOWRY O H. Distribution of guanine deaminase in mouse brain[J]. Journal of Neurochemistry, 1985, 44(6):1736-1740. DOI:10.1111/j.1471-4159.1985.tb07162.x.
[26] PALETZKI R F. Cloning and characterization of guanine deaminase from mouse and rat brain[J]. Neuroscience, 2002, 109(1): 15-26.DOI:10.1016/S0306-4522(01)00352-9.
[27] DAI X L, ZHUANG J, WU Y, et al. Identification of a flavonoid glucosyltransferase involved in 7-OH site glycosylation in tea plants (Camellia sinensis)[J]. Scientific Reports, 2017, 7(1): 735.DOI:10.1038/s41598-017-06453-z.
[28] 吴纬, 陈静娴, 宋宁, 等. 几种因素对重组大肠杆菌目标蛋白表达的影响[J]. 四川大学学报(自然科学版), 2002(增刊1): 155-157.
[29] MUKHOPADHYAY R, ROSEN B P. Arsenate reductases in prokaryotes and eukaryotes[J]. Environmental Health Perspectives,2002, 110(Suppl 5): 745-748. DOI:10.1289/ehp.02110s5745.
[30] 陈冠军, 邢金良, 商澎, 等. 外源基因在大肠杆菌中表达效率的影响因素[J]. 世界核心医学期刊文摘: 眼科学分册, 2004(3): 1121-1124.
[31] OLINS P O, LEE S C. Recent advances in heterologous gene expression in Escherichia coli[J]. Current Opinion in Biotechnology,1993, 4(5): 520-525. DOI:10.1016/0958-1669(93)90071-4 .
[32] 胡歆, 张玥. 黑茶渥堆中微生物与酶的变化及其代谢作用研究进展[C]//第十六届中国科协年会论文集. 昆明: 第十六届中国科协年会, 2014.
Cloning, Prokaryotic Expression and Functional Identification of Guanine Deaminase Genes from Saccharomyces cerevisiae and Escherichia coli
SUN Ying, PAN Si’an, LI Mengmeng, DENG Weiwei, ZHANG Zhengzhu*
(State Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, Hefei 230036, China)
Abstract: A novel pathway for the biosynthesis of caffeine with xanthine as substrate has been found in microbes, which is an effective way to improve the efficiency of caffeine synthesis from xanthine produced from guanine catalyzed by guanine deaminase. This study aimed to clone the guanine deaminase gene and to construct a prokaryotic expression system to synthesize xanthine eきciently. The two guanine deaminase genes gud1 and egud were amplified from Saccharomyces cerevisiae and Escherichia coli by PCR, respectively. The target fragments were inserted into the pMAL-c5X vector and then the recombinant plasmid transformed into E. coli BL21(DE3) to induce protein expression. The expressed products were determined by high performance liquid chromatography (HPLC). Both the recombinant vectors pMAL-gud1 and pMAL-egud had the capability of synthesizing xanthine, and the eきciency of xanthine synthesis with GUD1 was higher as compared to EGUD. This study can enrich the theory of black tea processing technology and provide a theoretical foundation for constructing engineered strains that are capable of producing caffeine.
Keywords: dark tea processing; caffeine; guanine deaminase; xanthine; biosynthesis; prokaryotic expression
SUN Ying, PAN Si’an, LI Mengmeng, et al. Cloning, prokaryotic expression and functional identification of guanine deaminase genes from Saccharomyces cerevisiae and Escherichia coli[J]. Food Science, 2018, 39(18): 139-144. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201818022. http://www.spkx.net.cn
孙莹, 潘思安, 李萌萌, 等. 酿酒酵母和大肠杆菌鸟嘌呤脱氨酶基因的克隆、原核表达及活性测定[J]. 食品科学, 2018,39(18): 139-144. DOI:10.7506/spkx1002-6630-201818022. http://www.spkx.net.cn
文章编号: 1002-6630(2018)18-0139-06
引文格式:
中图分类号: Q786
文献标志码: A
*通信作者简介: 张正竹(1969—),男,教授,博士,主要从事茶叶化学与加工、食品风味化学研究。E-mail:zzz@ahau.edu.cn
DOI: 10.7506/spkx1002-6630-201818022
基金项目: 教育部长江学者与创新团队滚动支持项目(IRT_15R01);国家自然科学基金面上项目(31570692)
第一作者简介: 孙莹(1993—),女,硕士研究生,主要从事茶叶生物化学及咖啡碱从头合成研究。E-mail:15720358sy@ahau.edu.cn
收稿日期: 2017-08-11