表1 样品18S rRNA测序情况及各分类地位数量
Table 1 18S rRNA read counts and number of identifiable units at different taxonomical levels

注:计算每个样品发现物种数和香农指数时,样品的测序量均为30 810 条序列。
王玉荣 1 ,代凯文 2 ,沈 馨 1 ,董 蕴 1 ,周书楠 1 ,郭 壮 1, *
(1.湖北文理学院化学工程与食品科学学院,鄂西北传统发酵食品研究所,湖北 襄阳 441053;2.当阳市食品药品监督管理局,湖北 当阳 444100)
摘 要: 采用MiSeq高通量测序技术对鲊广椒真菌微生物的群落结构进行解析的基础上,使用电子舌对不同样品各滋味指标的相对强度进行测定,最终对真菌多样性与鲊广椒滋味品质进行关联性分析。结果发现,在门水平上,Ascomycota(子囊菌门)的平均相对含量高达99.97%。在属水平上,相对含量大于1.0%的真菌属分别为Candida(念珠菌属)、Eurotium(曲霉菌属)、Pichia(毕赤酵母属)、Fusarium(镰刀霉属)、Galactomyces(地霉属)、Cladosporium(分枝孢子菌属)、Debaryomyces(德巴利氏酵母属)和Guehomyces(久浩酵母属),其平均相对含量分别为56.63%、10.12%、11.19%、2.67%、2.48%、2.21%、1.50%和1.01%。经皮尔森相关性分析发现优势真菌属与鲊广椒的苦味、涩味、后味A和后味B呈正相关。由此可见,真菌含量过高不利于鲊广椒滋味品质的形成。
关键词: 鲊广椒;高通量测序;电子舌;多样性;品质
作为我国特色传统发酵食品,湖北宜昌地区制作的鲊广椒以大米面、鲜红辣椒和食盐为主要原料,室温下发酵15~20 d而成,具有酸辣适口、滋味鲜香的特点。鲊广椒的发酵通常在倒扣于水盆的坛子中完成,同时坛口会塞入半坛稻草以防止原料浸入水中 [1] 。虽然发酵过程均在密封环境中完成 [2] ,但发酵初始阶段坛中存在的一定量空气,为专性好氧真菌的生长提供了必要的氧气。此外,鲊广椒的制作环境相对开放,加之环境中存在少量兼性厌氧和微好氧真菌,因而发酵好的鲊广椒产品中存在一定量的真菌,而该部分真菌的存在可能会对产品品质及使用安全性产生一定的影响,所以对鲊广椒中真菌多样性进行评价是极为必要的。
以Illumina MiSeq测序平台为代表的第2代测序技术具有通量高和检测速度快的特点 [3] ,通过为每个样品在测序前加上一段被称作样品特异性条形码或者标签的核苷酸序列,实现了多样本的平行实验 [4] ,解决了传统指纹图谱技术不能进行关联分析的缺点 [5] 。近年来,该技术被广泛应用于酸奶 [6] 、腊肠 [7] 、葡萄酒 [8] 、窖泥 [9] 和泡菜 [10] 等发酵食品微生物多样性研究和特异菌种的检测中,为全面和深入研究不同发酵食品基质中微生物分子生态学提供了新的技术手段。电子舌实现了食品酸味、苦味、涩味、鲜味、咸味、甜味、苦味回味、涩味回味和鲜味回味的数字化评价,具有结果稳定且受外界影响小的优点 [11] ,目前已经广泛应用于米酒 [12] 、啤酒 [13] 、水产品 [14] 和醋 [15] 等发酵食品的滋味品质评价中。
本研究从湖北省宜昌市当阳地区农户家中采集了10 个鲊广椒样品,首先采用MiSeq高通量测序技术对其真菌微生物多样性进行评价,继而使用电子舌系统对各滋味指标相对强度进行测定,最终对真菌多样性与鲊广椒滋味品质进行了关联性分析,以期对后续鲊广椒产品品质改良及安全性评价提供理论支持。
鲊广椒采集自湖北省当阳市草埠湖镇和玉泉办事处10 个农户家。
QIAGEN DNeasy mericon Food Kit食品DNA基因组提取试剂盒 德国QIAGEN公司;5×TransStart TM FastPfu Buffer、FastPfu Fly DNA Polymerase、dNTPs Mix北京全式金生物技术有限公司;DNA 1000试剂盒美国Agilent公司;味觉标准溶液、阴离子和阳离子溶液、内部溶液及参比溶液 日本Insent公司。
MiSeq高通量测序平台 美国Illumina公司;R920机架式服务器 美国Dell公司;SA 402B电子舌(配备AAE、CT0、CA0、AE1、C00和GL1测试传感器)日本Insent公司;2100芯片生物分析仪 美国Agilent公司;5810R台式高速冷冻离心机 德国Eppendorf公司;DYY-12电泳仪 北京六一仪器厂;ND-2000C微量紫外分光光度计 美国Nano Drop公司;UVPCDS8000凝胶成像分析系统 美国Bio-Rad公司;vetiri梯度基因扩增仪 美国AB公司。
1.3.1 样品微生物宏基因组DNA提取
取1.0 g鲊广椒,参照QIAGEN DNeasy mericon Food Kit试剂盒说明书进行DNA提取,检测合格后的DNA样品置-20 ℃暂存备用。
1.3.2 真菌18S rRNA V 4 ~V 5 区聚合酶链式反应(polymerase chain reaction,PCR)扩增
正向引物为SSU0817F(5’-TTAGCATGGAA TAATRRAATAGGA-3’),反向引物为SSU1196R(5’-TCTGGACCTGGTGAGTTTCC-3’) [16] ,其中在正向引物中加入7 个核苷酸标签(barcode)。PCR扩增体系为:4 μL 5×PCR缓冲液,2 μL 2.5 mmol/L dNTPs mix,0.8 μL 5 μmol/L正向引物,0.8 μL 5 μmol/L反向引物,0.4 μL 5 U/μL DNA聚合酶,10 ng DNA模板,体系用ddH 2 O补充至20 μL。PCR扩增条件为:95 ℃ 3 min;95 ℃ 30 s,55 ℃ 30 s,72 ℃ 45 s,30 个循环;72 ℃ 10 min。
1.3.3 样品平衡及MiSeq高通量测序
将10 个经1.0%琼脂糖凝胶电泳检验合格的扩增产物按照100 nmol/L浓度进行稀释混匀后,送至上海美吉生物医药科技有限公司使用MiSeq高通量测序平台进行测序。1.3.4 序列质控
根据成对序列之间的重叠(overlap)关系,将下机的双端序列数据拼接(merge)成一条序列。在序列拼接过程中根据以下标准进行质控:overlap区的碱基数需大于10 bp;最大错配比率需小于等于0.2;拼接好的序列需无barcode碱基错配;引物碱基错配数需小于等于2 bp。
序列拼接后,依照barcode信息将下机序列划分到各样品,对序列方向进行校正后切掉barcode和引物,进而得到高质量的序列。若切掉barcode和引物后的序列其碱基数小于50 bp,则该序列亦予以切除。
1.3.5 生物信息学分析
质控合格后的序列,采用QIIME(v1.70)平台 [17] 进行真菌微生物物种分析和多样性评价 [14] 。主要的处理流程为:1)采用PyNAST校准并把序列排齐 [18] ;2)首先在100%相似性下进行UCLUST归并 [19] ,建立无重复的单一的18S rRNA V 4 ~V 5 区序列集,继而在97.0%相似性下进行序列归并,构建分类操作单元(operational taxonomic units,OTU)矩阵;3)应用ChimeraSlayer去除含有嵌合体序列的OTU序列 [20] ;4)使用SILVA数据库 [21] 进行序列同源性比对,在门、纲、目、科和属水平上对其分类学地位进行明确;5)使用FastTree软件,绘制基于OTU代表序列的系统发育进化树 [22] ;6)采用发现物种数和香农指数(Shannon-Wiener index)等α多样性指标分别对单个样品真菌微生物的丰富度和多样性进行评价,同时,采用稀疏曲线和香农指数曲线对测序深度是否满足后续分子生物学分析进行评估 [23] ;7)分别基于UniFrac距离 [24] 进行主坐标分析和非加权组平均(unweighted pair-group method with arithmetic means,UPGMA)法聚类分析,进而完成不同样品间真菌微生物的β多样性分析。
1.3.6 核酸登录号
本研究中所有序列数据已提交至MG-RAST数据库,登录号为mgp81949。
1.3.7 基于电子舌技术鲊广椒滋味品质的评价
称取10 g鲊广椒样品于190 mL蒸馏水中,3 000 r/min离心10 min后取上清液过滤,按照下述步骤对鲊广椒样品酸、苦、涩、咸、鲜等基本味及涩、苦和鲜味回味进行测定 [25] :1)将C00和AE1传感器浸泡在阳离子溶液中,CA0、CT0和AAE传感器浸泡在阴离子溶液中,5 个传感器浸泡时间均为90 s,以去除传感器上的吸附物;2)5 个传感器在参比溶液1和2中分别洗涤120 s后,在参比溶液3中浸泡30 s,测得参比溶液的电势值V r ;3)5 个传感器在某鲊广椒样品中浸泡30 s,测得电势值V s ,通过计算V s -V r 值即可得到样品酸、苦、涩、咸和鲜味5 个基本味的相对强度值;4)传感器C00、AE1和AAE于参比溶液4和5中分别洗涤3 s后,于参比溶液6中浸泡30 s,测得电势V r ’,通过计算V r ’-V r 值即可得到样品后味A(涩的回味)、后味B(苦的回味)和丰度(鲜的回味)的相对强度值。每个样品重复测定4 次,选后3 次纳入数据分析。
GL1传感器在阴离子溶液中浸泡90 s后,参照上述程序单独对鲊广椒样品的甜味进行测定。每个样品重复测定5 次,选后3 次纳入数据分析。
1.3.8 多元统计学分析
使用皮尔森相关性分析法对平均相对含量大于1.0%的真菌属之间的相关性进行计算,并采用热图对结果进行展示;亦使用皮尔森相关性分析法对鲊广椒中优势真菌属和电子舌各滋味指标相对强度之间的相关性进行计算,同时选取相关系数绝对值大于0.5的指标,采用Cytoscape软件(v3.5.1)进行相关性网络图绘制。使用Origin 8.5软件(OriginLab Corp,MA,USA)和R软件(v3.3.2)进行作图。
在提取鲊广椒样品宏基因组DNA的基础上,本研究采用MiSeq高通量测序技术,对鲊广椒样品中真菌微生物多样性进行了评价。样品18S rRNA测序情况及各分类地位数量如表1所示。
表1 样品18S rRNA测序情况及各分类地位数量
Table 1 18S rRNA read counts and number of identifiable units at different taxonomical levels
注:计算每个样品发现物种数和香农指数时,样品的测序量均为30 810 条序列。
由表1可知,10 个鲊广椒样品共产生了376 256 条高质量的18S rRNA序列,平均每个样品产生37 626 条。本研究采用两步UCLUST法进行OTU的划分,根据序列100%相似性聚类分析后,得到69 496 条代表性序列,继而经过序列97.0%相似性聚类分析后,得到5 457 个OTU,经嵌合体检查后去除14 个OTU,还剩余5 443 个OTU进行后续分析。本研究有18.45%的OTU和3.91%的序列(范围为0%~14.11%,标准差为5.15%)不能鉴定到属水平。
采用稀疏曲线和香农指数曲线,本研究进一步对现有测序深度是否满足后续分子生物学分析进行了评估,其结果如图1所示。
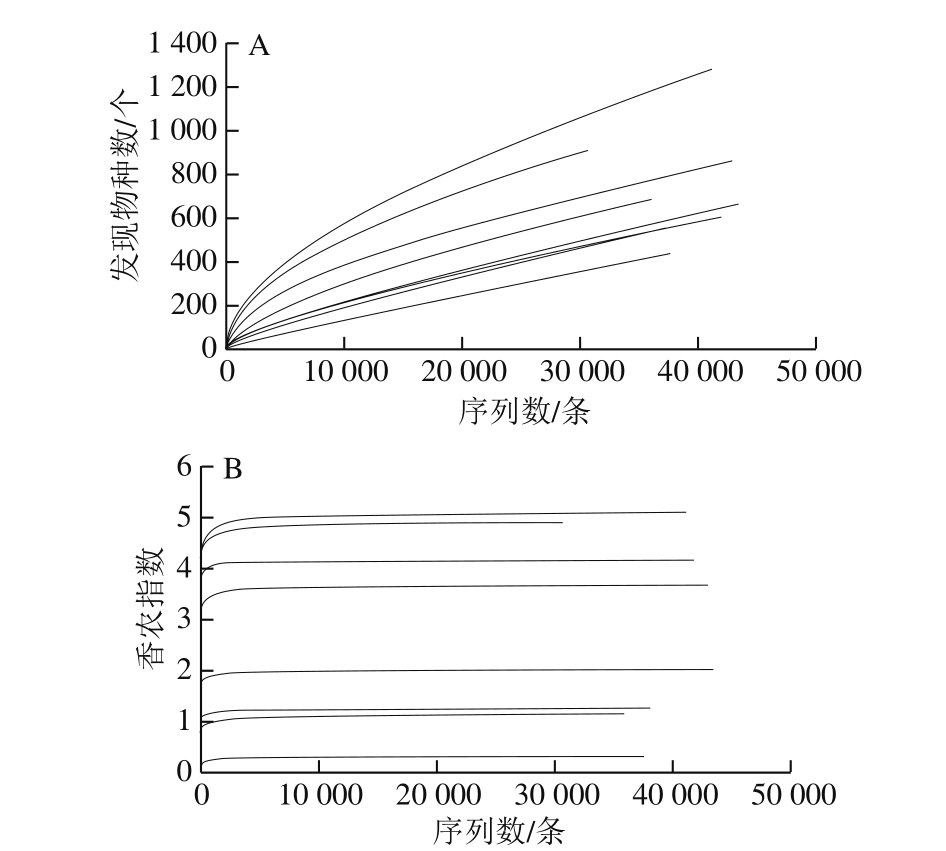
图1 稀疏曲线图(A)和香农指数图(B)
Fig. 1 Rarefaction analysis (A) and Shannon diversity estimates (B)
由图1可知,随着测序深度的增加,鲊广椒样品中发现真菌物种数的数量亦随之增大且稀疏曲线尚未进入平台期,而香农指数曲线已完全达到平衡状态。由此可见,随着测序深度的增加虽然有新的真菌种系型可能被发现,但本研究已经完全捕获到了鲊广椒样品中真菌类群的多样性信息,因而本研究的测序深度可以满足后续生物信息学分析要求 [26] 。
在门水平上,鲊广椒样品中微生物主要隶属于7 个真菌门,其中Ascomycota(子囊菌门)的平均相对含量高达99.97%。在属水平上,共发现135 个真菌属,鲊广椒中优势真菌属相对含量的比较分析如图2所示。

图2 鲊广椒中优势真菌属相对含量的比较分析
Fig. 2 Relative abundances of the major fungal genera in Zhaguangjiao samples
由图2可知,鲊广椒中隶属于Ascomycota(子囊菌门)且平均相对含量大于1.0%的真菌属及其相对含量分别为:Candida(念珠菌属,56.63%)、Eurotium(曲霉菌属,10.12%)、Pichia(毕赤酵母属,11.19%)、Fusarium(镰刀霉属,2.67%)、Galactomyces(地霉属,2.48%)、Cladosporium(分枝孢子菌属,2.21%)和Debaryomyces(德巴利氏酵母属,1.01%)。此外,还有隶属于Basidiomycota(担子菌门)的Guehomyces(久浩酵母属),其平均相对含量为1.50%。由此可见,当阳地区鲊广椒主要是由若干个隶属于Ascomycota(子囊菌门)已知的优势真菌属组成,其平均累计含量为86.32%。Candida(念珠菌属)和Pichia(毕赤酵母属)在10 个样品中均存在,其平均累计含量分别为67.82%。平均相对含量大于1.0%的真菌属相关性的热图如图3所示。
由图3可知,Galactomyces(地霉属)与Candida(念珠菌属)呈显著负相关(R=-0.673,P<0.05),与Eurotium(曲霉菌属)呈极显著正相关(R=0.893,P<0.01),Guehomyces(久浩酵母属)与Fusarium(镰刀霉属)呈极显著正相关(R=0.867,P<0.01),Debaryomyces(德巴利氏酵母属)与Fusarium(镰刀霉属,R=0.667)和Guehomyces(久浩酵母属,R=0.719)均呈显著正相关(P<0.05)。本研究进一步在OTU水平上对鲊广椒真菌微生物的多样性进行分析,OTU在10 个样品中出现次数统计如图4所示。
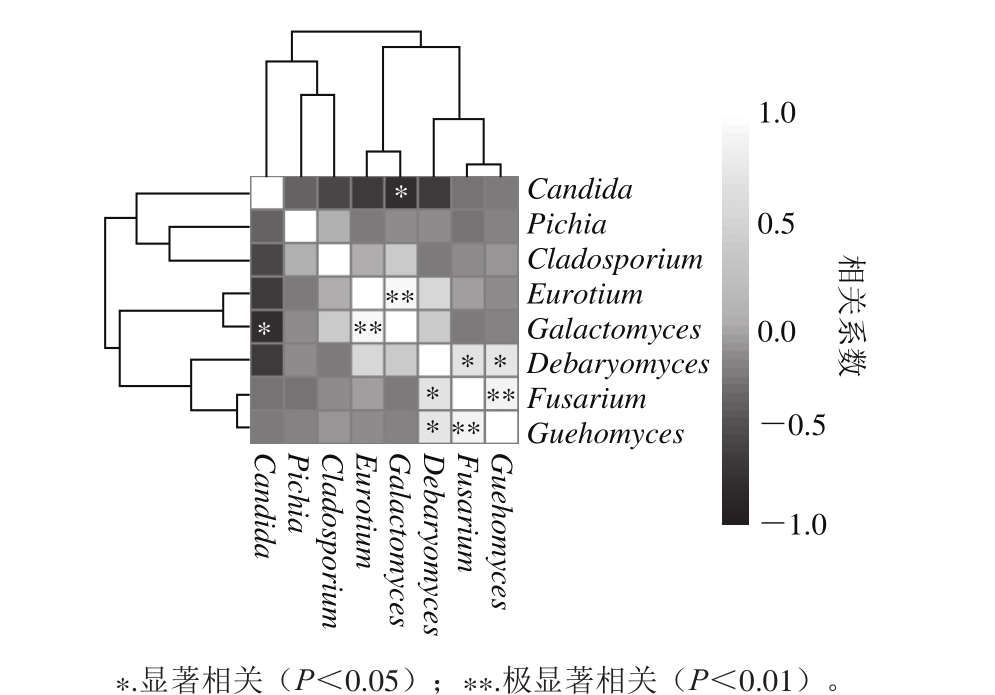
图3 平均相对含量大于1.0%的真菌属相关性的热图
Fig. 3 Heat map of correlation among the fungal genera with relative abundances more than 1.0%
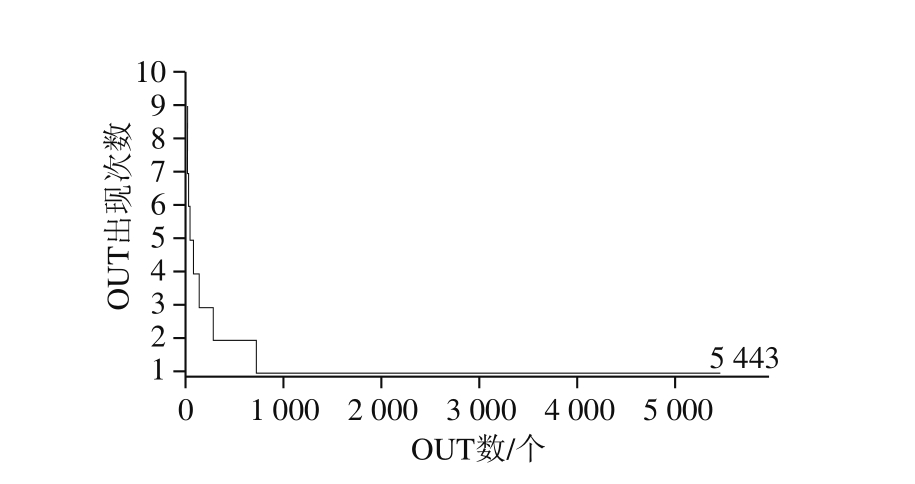
图4 OTU在10 个样品中出现次数统计
Fig. 4 Distribution of OTU as a function of their prevalence in the 10 samples
由图4可知,纳入本研究的样品共产生5 443 个OTU,然而在10 个样品中仅出现1 次的OTU有4 715 个,占OTU总数的86.63%,所包含序列数为13 070 条,仅占所有质控后合格序列数的3.47%。10 个样品没有共有的OTU,仅有5 个OTU存在9 个样品中,其包含序列数为203 208 条,高达所有质控后合格序列数的54.01%。由此可见,在OTU水平上,虽然若干鲊广椒样品存在大量的核心真菌菌群,但某些样品间微生物群落结构的构成存在较大差异。
通过上述分析,本研究发现虽然多数鲊广椒样品存在大量的核心真菌菌群,但某些样品间微生物群落结构的构成存在较大差异,因而本研究进一步采用基于OTU水平的加权UniFrac距离主坐标分析和UPGMA聚类分析,对10 个鲊广椒样品的β多样性进行了分析,基于分类操作单元的加权UniFrac距离的主坐标分析如图5所示。

图5 基于分类操作单元的加权UniFrac距离的主坐标分析
Fig. 5 Principal coordinate analysis of OTUs based on weighted UniFrac distance
由图5可知,10 个鲊广椒样品呈现明显的分离趋势,其中DY1、DY2、DY5和DY8,DY4、DY6、DY7和DY9,DY3和DY10分别呈现出聚类趋势。基于分类操作单元的加权UniFrac距离的UPGMA聚类分析如图6所示。

图6 基于分类操作单元的加权UniFrac距离的UPGMA聚类分析
Fig. 6 UPGMA clustering analysis of OTUs based on weighted UniFrac distance
由图6可知,10 个样品整体上可以分为3 个聚类,其中聚类I由样品DY4、DY6、DY7和DY9构成,聚类II由样品DY1、DY2、DY5和DY8构成,聚类III由DY3和DY10构成,这与基于分类操作单元的加权UniFrac距离的主坐标分析结果一致,亦进一步证实了虽然若干鲊广椒样品存在大量的核心真菌菌群,但某些样品间其微生物群落结构构成存在较大的差异。通过Krushkal-Wallis检验发现,隶属于不同聚类的样品除Eurotium(曲霉菌属)和Guehomyce(久浩酵母属)外,其他优势真菌属差异均不显著(P>0.05)。

图7 鲊广椒各滋味指标相对强度值的箱形图(n=10)
Fig. 7 Box plot of relative intensity of each taste index (n = 10)
在采用MiSeq高通量测序技术对真菌微生物多样性进行揭示的基础上,使用电子舌对鲊广椒样品的滋味品质进行评价,在此基础上构建了优势真菌属与产品滋味品质相关性的网络图。鲊广椒各滋味指标相对强度值的箱形图如图7所示。
由图7可知,鲊广椒样品在酸味上的差异最大,其次为咸味、甜味和鲜味,而在苦味、涩味、后味A(涩的回味)、后味B(苦的回味)和丰度(鲜的回味)指标上的差异较小。鲊广椒优势真菌属和滋味物质相关性的网络图如图8所示。
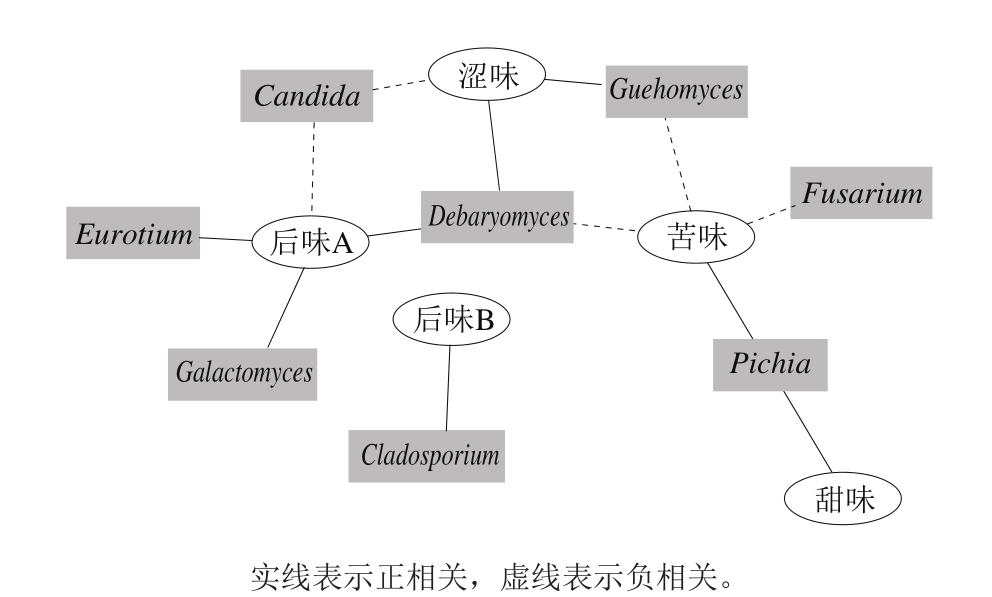
图8 鲊广椒优势真菌属和滋味物质相关性的网络图
Fig. 8 Correlation network diagram of major fungal genera and taste attributes in Zhaguangjiao samples
由图8可知,Pichia(毕赤酵母属)与鲊广椒苦味呈正相关;Debaryomyces(德巴利氏酵母属)和Guehomyce(久浩酵母属)与鲊广椒涩味呈正相关;Debaryomyces(德巴利氏酵母属)、Galactomyces(地霉属)和Eurotium(曲霉菌属)与鲊广椒后味A(涩味的回味)呈正相关;Galactomyces(地霉属)与后味B(苦味的回味)呈正相关。苦味、涩味、后味A和后味B均为鲊广椒的缺陷型指标,由此可见,优势真菌属多与鲊广椒品质的缺陷性指标相关,真菌含量过高可能不利于产品滋味品质的形成。
近年来出现的以Illumina MiSeq为代表的第2代高通量测序技术可以全面、客观、无偏地了解某一微生态系统中微生物群落结构,具有通量高、检测速度快和检测结果准确的特点 [27] 。本研究采用该技术对当阳地区鲊广椒中真菌微生物多样性进行了评价,结果发现念珠菌属、曲霉菌属和毕赤酵母属为其优势真菌属,相对含量占到了真菌数的近八成。念珠菌是真菌中最常见的条件致病菌,曲霉菌属亦是引起多种物质霉腐的主要微生物之一,且鲊广椒样品中亦存在隶属于镰刀菌属、地霉属和分枝孢子菌属的腐败真菌 [28] 。制作鲊广椒的发酵方式为厌氧发酵,然而发酵初始阶段坛中存在的空气及发酵后期居民常开坛取食鲊广椒,均为真菌的生长提供了必需的氧气,使鲊广椒受腐败真菌污染的机率大大增加。此外,制作鲊广椒的坛子常倒扣于水盆中并置于墙角或房檐下,用于密封的水亦常因家禽饮用或雨水溅入而发臭,这也增加了鲊广椒被腐败真菌污染的机会。由此可见,改善鲊广椒加工环境进而抑制腐败霉菌的生长,对保障鲊广椒的食用安全性具有积极的意义。
在采用MiSeq高通量测序技术对真菌微生物多样性进行评价的基础上,本研究使用电子舌系统对各滋味指标相对强度进行测定,结果发现鲊广椒样品在酸味上的差异最大,这可能与样品中乳酸菌含量不同有关 [29] ,因为除真菌外鲊广椒中亦存在大量细菌。此外,通过将真菌多样性与鲊广椒滋味品质进行关联性分析发现,优势真菌属多与鲊广椒品质的缺陷性指标相关,因而综上所述,当阳地区鲊广椒中真菌微生物主要隶属于念珠菌属、曲霉菌属和毕赤酵母属,且其含量过高可能不利于产品滋味品质的形成。此外,MiSeq高通量测序技术存在读长短的缺陷,因而二代测序只能在属的水平上研究样本中菌群的组成和变化,难以精确到种水平上 [30] ,因而在后续研究中积极引入新的测序技术并实现鲊广椒样品中活的真菌的绝对定量,对鲊广椒产品品质改良及安全性评价将具有积极的意义。
参考文献:
[1] 王巧碧, 王丹, 赵欠, 等. SDE和SPME法对鲊海椒发酵中香气组成的比较分析[J]. 食品科学, 2016, 37(4): 108-114. DOI:10.7506/spkx1002-6630-201604020.
[2] 葛平珍, 王丹, 周才琼. 不同淀粉源对鲊海椒发酵过程中功能成分的影响[J]. 食品科学, 2015, 36(21): 191-195. DOI:10.7506/spkx1002-6630-201521036.
[3] CAPORASO J G, LAUBER C L, WALTERS W A, et al. Ultra-highthroughput microbial community analysis on the Illumina HiSeq and MiSeq platforms[J]. The ISME Journal, 2012, 6(8): 1621-1624.DOI:10.1038/ismej.2012.8.
[4] ROH S W, KIM K H, NAM Y D, et al. Investigation of archaeal and bacterial diversity in fermented seafood using barcoded pyrosequencing[J]. The ISME Journal, 2010, 4(1): 1-16. DOI:10.1038/ismej.2009.83.
[5] KING J L, LARUE B L, NOVROSKI N M, et al. High-quality and high-throughput massively parallel sequencing of the human mitochondrial genome using the Illumina MiSeq[J]. Forensic Science International: Genetics, 2014, 12(10): 128-135. DOI:10.1016/j.fsigen.2014.06.001.
[6] 智楠楠. Illumina Miseq平台深度测定酸奶中微生物多样性[J].食品工业科技, 2016, 37(24): 78-82. DOI:10.13386/j.issn1002-0306.2016.24.007.
[7] JUSTYNA P, ANNALISA R, VINCENZA P, et al. Bacterial diversity in typical Italian salami at different ripening stages as revealed by high-throughput of 16S rDNA amplicons[J]. Food Microbiology,2015, 46(4): 342-356. DOI:10.1016/j.fm.2014.08.023.
[8] BOYNTON P J, GREIG D. Fungal diversity and ecosystem function data from wine fermentation vats and microcosms[J]. Data in Brief,2016, 8(12): 225-229. DOI:10.1016/j.dib.2016.05.038.
[9] HU X, DU H, REN C, et al. Illuminating anaerobic microbial community and cooccurrence patterns across a quality gradient in Chinese liquor fermentation pit muds[J]. Applied and Environmental Microbiology, 2016, 82(8): 2506-2515. DOI:10.1128/AEM.03409-15.
[10] YANG H, WU H, GAO L, et al. Effects of Lactobacillus curvatus and Leuconostoc mesenteroides on Suan Cai fermentation in northeast China[J]. Journal of Microbiology and Biotechnology, 2016, 26(12):2148-2158. DOI:10.4014/jmb.1607.07010.
[11] KHAN M R R, KHALILIAN A, KANG S W. A high sensitivity IDC-electronic tongue using dielectric/sensing membranes with solvatochromic dyes[J]. Sensors, 2016, 16(5): 668-687. DOI:10.3390/s16050668.
[12] 王玉荣, 张俊英, 胡欣洁, 等. 湖北孝感和四川成都地区来源的酒曲对米酒滋味品质影响的评价[J]. 食品科学, 2015, 36(16): 207-210.DOI:10.7506/spkx1002-6630-201516038.
[13] GUTIÉRREZ J M, HADDI Z, AMARI A, et al. Hybrid electronic tongue based on multisensor data fusion for discrimination of beers[J]. Sensors and Actuators B: Chemical, 2013, 177(2): 989-996.DOI:10.1016/j.snb.2012.11.110.
[14] 张晶晶, 顾赛麒, 丁玉庭, 等. 电子舌在中华绒螯蟹产地鉴别及等级评定的应用[J]. 食品科学, 2015, 36(4): 141-146. DOI:10.7506/spkx1002-6630-201504027.
[15] LIU M, WANG M, WANG J, et al. Comparison of random forest,support vector machine and back propagation neural network for electronic tongue data classification: application to the recognition of orange beverage and Chinese vinegar[J]. Sensors and Actuators B:Chemical, 2013, 177(2): 970-980. DOI:10.1016/j.snb.2012.11.071.
[16] WU H, ZHANG S, MA Y, et al. Comparison of microbial communities in the fermentation starter used to brew Xiaoqu liquor[J]. Journal of the Institute of Brewing, 2017, 123(1): 113-120. DOI:10.1002/jib.388.[17] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods, 2010, 7(5): 335-336. DOI:10.1038/nmeth.f.303.
[18] CAPORASO J G, BITTINGER K, BUSHMAN F D, et al. PyNAST: a flexible tool for aligning sequences to a template alignment[J].Bioinformatics, 2010, 26(2): 266-267. DOI:10.1093/bioinformatics/btp636.
[19] EDGAR R C. Search and clustering orders of magnitude faster than BLAST[J]. Bioinformatics, 2010, 26(19): 2460-2461. DOI:10.1093/bioinformatics/btq461.
[20] HAAS B J, GEVERS D, EARL A M, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J]. Genome Research, 2011, 21(3): 494-504.DOI:10.1101/gr.112730.110.
[21] QUAST C, PRUESSE E, YILMAZ P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools[J]. Nucleic Acids Research, 2012, 41(1): 590-596. DOI:10.1093/nar/gks1219.
[22] PRICE M N, DEHAL P S, ARKIN A P. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix[J]. Molecular Biology and Evolution, 2009, 26(7): 1641-1650.DOI:10.1093/molbev/msp077.
[23] ZHANG J, GUO Z, XUE Z, et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles,geography and ethnicities[J]. The ISME Journal, 2015, 9(9) : 1-12.DOI:10.1038/ismej.2015.11.
[24] LOZUPONE C, KNIGHT R. UniFrac: a new phylogenetic method for comparing microbial communities[J]. Applied and Environmental Microbiology, 2005, 71(12): 8228-8235. DOI:10.1128/AEM.71.12.8228-8235.2005.
[25] 郭壮, 汤尚文, 王玉荣, 等. 基于电子舌技术的襄阳市售米酒滋味品质评价[J]. 食品工业科技, 2015, 36(15): 289-293. DOI:10.13386 /j.issn1002-0306.2015.15.052.
[26] WANG T, CAI G, QIU Y, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers[J]. The ISME Journal, 2012, 6(2): 320-329. DOI:10.1038/ismej.2011.109.
[27] QUAIL M A, SMITH M, COUPLAND P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers[J]. BMC Genomics, 2012,13(1): 341. DOI:10.1186/1471-2164-13-341.
[28] QUAN L S, TSAO M. Biocontrol of spoilage yeasts and moulds by Williopsis saturnus var. saturnus in yoghurt[J]. Nutrition & Food Science, 2010, 40(2): 166-175. DOI:10.1108/00346651011029192.
[29] CORSETTI A, SETTANNI L. Lactobacilli in sourdough fermentation[J]. Food Research International, 2007, 40(5): 539-558.DOI:10.1016/j.foodres.2006.11.001.
[30] KOZICH J J, WESTCOTT S L, BAXTER N T, et al. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform[J].Applied and Environmental Microbiology, 2013, 79(17): 5112-5120.DOI:10.1128/AEM.01043-13.
Characterization of Fungal Microflora and Its Influence on Taste Quality of Zhaguangjiao, a Chinese Traditional Fermented Chili Product
WANG Yurong 1 , DAI Kaiwen 2 , SHEN Xin 1 , DONG Yun 1 , ZHOU Shunan 1 , GUO Zhuang 1, *
(1. Northwest Hubei Research Institute of Traditionally Fermented Food, School of Chemical Engineering and Food Science,Hubei University of Arts and Science, Xiangyang 441053, China;2. Dangyang Municipal Food and Drug Administration, Dangyang 444100, China)
Abstract: The fungal community structure in Zhaguangjiao was analyzed by MiSeq high throughput sequencing and the relative intensity of various taste attributes of Zhaguangjiao samples was determined using an electronic tongue. Furthermore,the correlation between the fungal diversity and taste quality of was evaluated. The results showed that Ascomycota was the most dominant fungal phyla with a relative abundance of 99.97%. Candida, Eurotium, Pichia, Fusarium, Galactomyces,Cladosporium, Guehomyces, and Debaryomyces each accounted for more than 1.0% of total sequences at the genus level,with a relative abundance of 56.63%, 10.12%, 11.19%, 2.67%, 2.48%, 2.21%, 1.50% and 1.01%, respectively. The results of Pearson correlation analysis indicated that there were positive correlations among the relative abundance of the dominant fungal genera and the defective indexes of taste quality. Thus, we can consider that the fungal microbiota played inactive roles in the taste quality formation of Zhaguangjiao.
Keywords: Zhaguangjiao; high throughput sequencing; electronic tongue; diversity; quality
WANG Yurong, DAI Kaiwen, SHEN Xin, et al. Characterization of fungal microflora and its influence on taste quality of Zhaguangjiao, a Chinese traditional fermented chili product[J]. Food Science, 2018, 39(18): 173-178. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201818027. http://www.spkx.net.cn
王玉荣, 代凯文, 沈馨, 等. 鲊广椒真菌多样性及其对滋味品质影响的评价[J]. 食品科学, 2018, 39(18): 173-178.DOI:10.7506/spkx1002-6630-201818027. http://www.spkx.net.cn
文章编号: 1002-6630(2018)18-0173-06
引文格式:
中图分类号: TS201.4
文献标志码: A
*通信作者简介: 郭壮(1984—),男,副教授,博士,研究方向为食品生物技术。E-mail:guozhuang1984@163.com
DOI: 10.7506/spkx1002-6630-201818027
基金项目: 湖北文理学院教师科研能力培育基金项目(2017kypy051)
第一作者简介: 王玉荣(1993—),女,硕士,研究方向为食品生物技术。E-mail:wangyurong1993@163.com
收稿日期: 2017-08-20