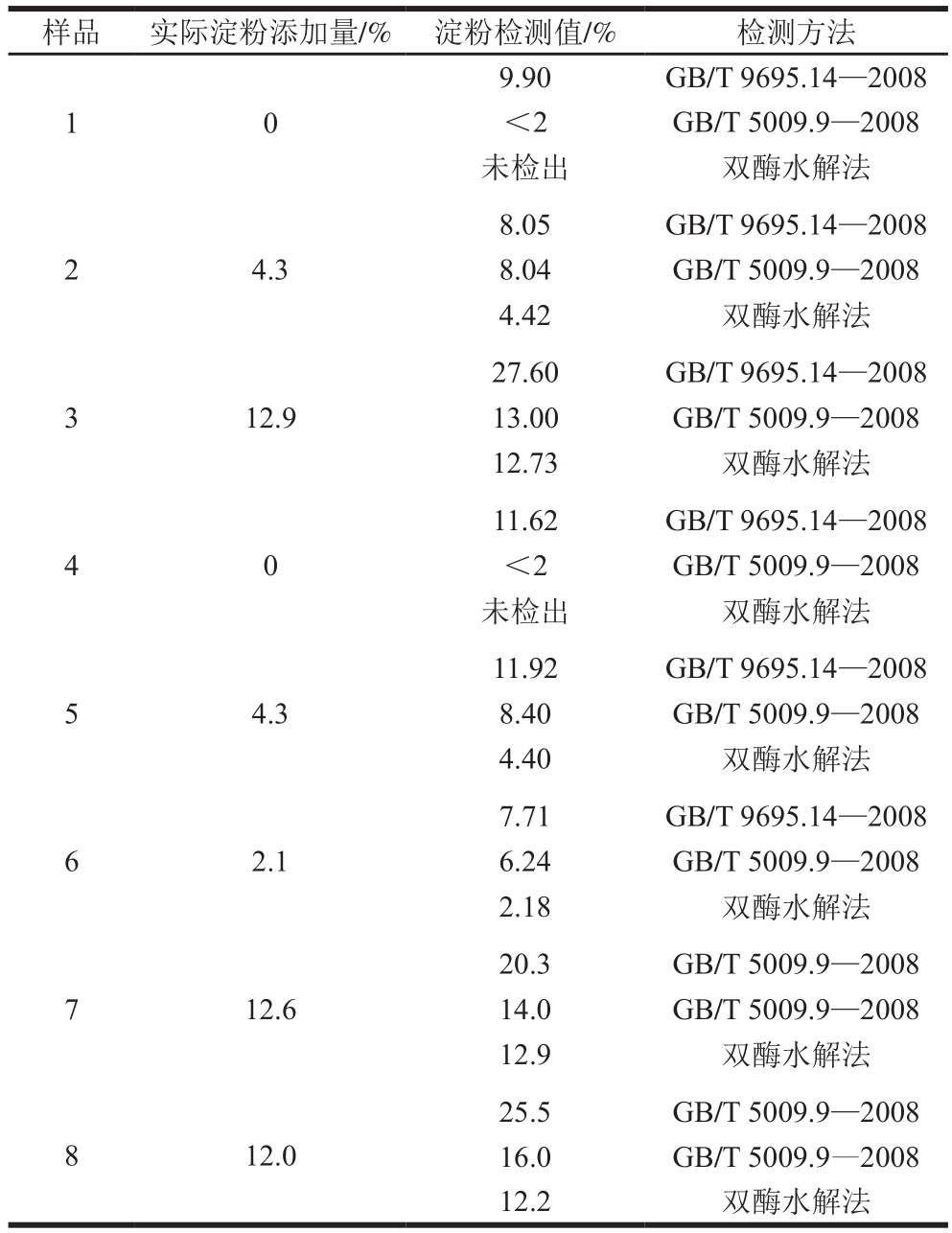
表1 不同检测方法对相同样品的检测
Table 1 Comparison of starch contents of known samples determined by different methods
翁晨辉 1,2 ,陈小珍 1,2, *,林卫星 3 ,吴 红 4 ,金志瑜 3
(1.浙江工业大学化学工程学院,浙江 杭州 310014;2.浙江省产品质量安全检测研究院,浙江 杭州 310013;3.浙江迪恩安正检测技术有限公司,浙江 杭州 310024;4.杭州唯新食品有限公司,浙江 杭州 310052)
摘 要: 建立超声辅助双酶水解法联合高效液相色谱测定肉松制品中淀粉含量的检测方法。采用超声辅助双酶水解法水解肉松中的淀粉后,利用高效液相色谱法对水解液中的葡萄糖含量进行测定,并换算为淀粉含量,采用C 18 色谱柱(4.6 mm×250 mm,5 μm),以乙腈-20 mmol/L乙酸铵溶液(22∶78,V/V)为流动相,以245 nm为分析波长,葡萄糖在5~50 mg/L质量浓度范围内线性良好,相关系数r大于0.999。本实验采用耐高温α-淀粉酶与淀粉葡萄糖苷酶酶解淀粉,该方法仪器检出限为0.4 μg/mL,方法检出限为0.16%。通过肉松水解液加标验证,平均回收率为88.0%~96.6%,相对标准偏差小于5%(n=6)。与国家标准方法相比,该方法重复性好、回收率高,适用于各种肉松中淀粉含量的测定。
关键词: 高效液相色谱;水解;肉松;淀粉;检测
淀粉是由葡萄糖聚合而成的可食用、可被生物降解的天然高分子化合物 [1] 。它是植物光合作用的产物,广泛存在于不同植物的种子、块茎和块根等组织中,是全球膳食结构中最丰富的营养来源 [2] 。淀粉可应用于造纸业、纺织业、医药产业、食品加工业等 [3-6] 。在食品加工业中,淀粉具有改善食品质量和稠度的作用,在肉制品中,淀粉可作为增稠剂、赋形剂、乳化剂、填充剂等,以改善肉制品持水性、黏着性和组织形态等物理性质,在肉松中淀粉可以起到改善组织状态、提高成品率、节约生产成本等作用 [7-13] 。
淀粉作为食品添加剂,其添加量的多少是鉴别肉制品质量高低及产品合格与否的指标 [14] 。近年来,我国一些肉制品加工企业为了追求经济利益滥用淀粉,即使用劣质淀粉、过量使用淀粉或在不得添加淀粉的制品中添加淀粉,达到非法盈利的目的。按照国家标准规定,干肉制品中,肉脯和肉干制品不得添加淀粉,肉松制品中,油酥肉松和肉松的淀粉添加量应小于2 g,肉粉松的添加量应小于30%。2017年6月23日,GB 5009.9—2016《食品中淀粉的测定》 [15] 代替GB/T 9695.14—2008《肉制品 淀粉含量测定》 [16] 和GB/T 5009.9—2008《食品中淀粉的测定》 [17] ,但由于GB 5009.9—2016将现行有效的2 种检测方法合二为一,根本问题并没有得到妥善解决。在肉松制品检测过程中,时常出现假阳性或检测结果与实际添加量出入较大的情况,若采用不同国家标准方法对同一种样品进行检测,检测结果差别较大,甚至出现采用一种国家标准方法检测结果为“合格”,另一种为“不合格”的情况,不仅如此,该国家标准方法还存在一定局限性,即不适用于同时含有经水解也能产生还原糖的其他添加物的淀粉测定,也不适用于大批量样品的测试,给检验和监督工作带来一定困难,也让不法商贩有机可乘,给社会带来负面影响 [18-19] 。因此,作为肉松产品评定的重要指标,淀粉的准确测定至关重要,其检测技术的开发将对日常监管起到推动作用。
本实验拟利用超声辅助耐高温α-淀粉酶和淀粉葡萄糖苷酶水解肉松中的淀粉,通过高效液相色谱法检测水解液中葡萄糖含量,以此换算为淀粉含量。该方法在国内肉松行业淀粉指标的检测中鲜有报道,也未见相关的国家标准,本实验方法可有效规避现有国家标准方法的缺点,保证淀粉测定的准确性,且超声波可提高酶解效率,减少实验时间。国内外对淀粉的检测主要集中在滴定法、旋光法、比色法、碘显色法等,但液相色谱法较这些方法具有高灵敏度、高准确性及高效性等优点,不仅可以应用于肉松制品,还可以推广至其他食品,因此采用高效液相色谱法是近年来的发展趋势。1-苯基-3-甲基-5-吡唑啉酮(3-methyl-1-phenyl-2-pyrazolin-5-one,PMP)柱前衍生化法被广泛应用于药物糖的分析和提取 [20-21] ,如当归 [22] 、七叶胆 [23] 、白沙蒿 [24] 、石莼 [25] 、决明子 [26] 等。该方法还可应用于其他植物和食品中多糖的检测。Dai Jun等 [27] 研究12 种单糖-PMP衍生物的分离,优化了流动相、pH值、反应时间等实验条件,并将该方法应用于盐藻多糖中单糖成分的分析。Sun Lijun等 [28] 利用PMP衍生化法和气相色谱-质谱确定苹果皮多糖中的单糖组成,并比较了这2 种方法的差异点。目前,关于PMP衍生化法测定肉制品中淀粉的报道较少。本研究建立高效液相色谱测定肉松中淀粉的分析方法,并采用该方法对9 个不同产品进行分析测定和比较,以期为肉松中淀粉的检测提供新方法,并为肉松的质量评价及监管部门提供技术支持。
肉松、肉绒等样品 市购。葡萄糖标准品(纯度≥99%) 质检科技股份有限公司;耐高温α-淀粉酶、PMP 阿拉丁试剂(上海)有限公司;淀粉葡萄糖苷酶 上海麦克林生化科技有限公司;乙腈(色谱纯)美国Tedia公司;0.45 μm微孔滤膜 上海兴亚净化材料厂;其他化学试剂均为国产分析纯。
BSA224S型电子分析天平 赛多利斯科学仪器(北京)有限公司;KQ-500DA型超声清洗器 昆山市超声仪器有限公司;LC-20A型高效液相色谱仪(附紫外检测器) 日本岛津公司;ST16R型高速冷冻离心机赛默飞世尔科技(中国)有限公司;TDL-80-2B型离心机 上海安亭科学仪器;Milli-Q超纯水系统 密理博中国有限公司;SHA-CA型水浴恒温振荡器 金坛市荣华仪器制造有限公司;HHS型电热恒温水浴锅 上海博迅实业有限公司医疗设备厂。
1.3.1 国家标准方法对比
GB 5009.9—2016第2法:称取2 g(精确到0.001 g)样品,置于放有慢速滤纸的漏斗中,用50 mL石油醚分5 次洗涤样品,残渣用150 mL 85%乙醇溶液分数次洗涤,滤干,用100 mL水洗涤残渣至250 mL锥形瓶中,加入30 mL盐酸(1∶1,V/V),置沸水浴中回流2 h。待试样冷却后,调节pH值至中性,加入200 g/L乙酸铅溶液和100 g/L硫酸钠溶液各20 mL,摇匀,定容至500 mL,过滤,弃去初滤液20 mL,滤液供测定。采用斐林试剂法测定滤液中的葡萄糖含量。
GB 5009.9—2016第3法:称取试样25 g(精确到0.01 g,淀粉含量约1 g),置于500 mL烧杯中,加入300 mL热氢氧化钾-乙醇溶液(5%),沸水浴加热1 h,不时搅拌。沉淀用80%热乙醇溶液洗涤数次后,用100 mL 1.0 mol/L盐酸溶液洗入250 mL烧杯中,沸水浴中水解2.5 h,不时搅拌。待试样冷却后,调节pH值约为6,加入106 g/L铁氰化钾溶液和220 g/L乙酸锌溶液各3 mL,用水定容至200 mL。摇匀,过滤,滤液中加入300 g/L氢氧化钠溶液1~2 滴。采用碘量法测定滤液中的葡萄糖含量。
1.3.2 双酶水解高效液相色谱法
1.3.2.1 样品水解
称取研磨均匀的样品1.000 g,置于50 mL离心管中,用50 mL石油醚分2 次洗除脂肪,氮吹干肉松层后再用50 mL超纯水分2 次洗去可溶性糖类,残留物加1 000 μL耐高温α-淀粉酶,并加入超纯水至15 mL,超声20 min,于100 ℃恒温水浴中糊化60 min,取出冷却至60 ℃,加入100 μL淀粉葡萄糖苷酶溶液,于65 ℃恒温水浴中糖化60 min,取出调节pH值至2.2,灭酶30 min,定容至20 mL。
1.3.2.2 衍生化处理
采用葡萄糖与PMP试剂衍生化法 [29-31] 。旋涡酶解液,离心后取上清液10 μL于10 mL离心管中,加入100 μL 0.5 mol/L PMP-甲醇溶液及100 μL 2 mol/L氨水溶液,于70 ℃水浴中反应30 min,取出氮吹干,加入2 mL超纯水和2 mL三氯甲烷,旋涡离心取上清液,重复提取2 次,上清液过0.45 μm滤膜后供高效液相色谱分析。
1.3.2.3 高效液相色谱分析条件
色谱柱:Kromasil 100-5C 18 (4.6 mm×250 mm,5 μm);流动相:乙酸铵-乙腈(78∶22,V/V)溶液;流速1.0 mL/min;柱温35 ℃;波长245 nm;进样量10 μL。
配制不同质量浓度的葡萄糖标准溶液,以葡萄糖质量浓度为横坐标,峰面积为纵坐标,绘制标准曲线。
如表1所示,同一种样品采用不同方法检测,结果差异明显,对产品合格与否的评定影响较大。样品3采用2 种国家标准方法测定,结果分别为27.60%和13.00%,两者结果相差52.9%,采用GB/T 9695.14—2008方法的结果接近限量值,若干扰物添加量大于现值或存在其他不确定因素,极有可能导致产品(淀粉实际添加值小于限量)淀粉含量超标。同一样品采用同一方法由不同的检测人员检测,其差异明显,如样品7和样品8所示,两者结果分别相差31.0%和37.3%。且采用GB/T 9695.14—2008和GB/T 5009.9—2008方法与淀粉实际添加量相差较大,而采用双酶水解与实际结果相近,由此表明超声辅助双酶水解法是一种可靠准确的方法,可以有效替代现有国家标准方法。
新实施的标准与原GB/T 5009.9—2008相比,主要增加了低含量样品和试剂空白测定,修改了第1法中的计算公式以及增加了第3法:肉制品中淀粉含量测定,即把原GB/T 9695.14—2008方法合并。
表1 不同检测方法对相同样品的检测
Table 1 Comparison of starch contents of known samples determined by different methods
由于GB 5009.9—2016第1法酶水解法与第2法酸水解法在实际样品的检测中,检测结果基本相同,且酶水解法重复性稍差 [32] ,所以现阶段实验室多采用第2法和第3法对肉松样品进行检测,故本实验将着重比较这2 种方法的差别。第2法和第3法的原理相似,都是将样品去脂及可溶性糖后,用酸水解,测定水解液中还原糖含量,并折算成淀粉。2 种方法的差别主要集中在称样量,除脂、除糖方法,以及水解方法、时间和测定方法,如表2所示。
表2 国家标准检测方法的差别
Table 2 Comparison of different starch detection procedures

由于肉类本身含有糖类物质,如葡萄糖、核糖、糖原等,它们以游离或结合的形式广泛存在于动物组织或组织液中,当称样量为25 g时,样品中的脂肪、蛋白质以及肉本身的含糖量较高,易产生误差。第3法以热氢氧化钾-乙醇溶液300 mL于沸水浴上加热1 h除去脂肪和蛋白质,后续用热乙醇除可溶性糖,在此过程中粗纤维以及其他糖类物质不溶解,将造成结果正误差。水解过程中,糖苷键水解是随机的,所以盐酸用量及酸水解时间是影响淀粉水解的主要因素,若酸用量少,水解时间短,则水解不完全。第3法加入的盐酸量(平均每克样品)较第2法多,且水解过程采用沸水浴水解2.5 h,该方法需不停搅拌,若不搅拌,淀粉将附着在烧杯上,造成水解不完全,第2法则采用冷凝回流2 h,水解过程更剧烈。还原糖的测定第3法采用碘量法,第2法采用斐林试剂法,斐林试剂法的指示剂为次甲基蓝,其颜色变化受反应液温度和持续沸腾加热时间影响,若滴定过程并没有完全在沸腾状态条件下进行,将导致滴定终点延后,一般沸腾时间短,消耗糖量多,沸腾时间长,消耗糖量少。且肉松样品多为深色样品,常导致滴定终点模糊不清,不同操作人员对终点判断也不同,最终造成误差。
双酶水解法具有取样量少、特异性强、检测效率高等优点,适用于大批量样品的检测。如表1所示,相同样品采用双酶水解法比采用国家标准方法更准确,含量更低,因为α-淀粉酶和淀粉葡萄糖苷酶体系对淀粉的水解具有高效性和专一性,经酸水解会产生还原性糖的干扰物质在双酶水解法中不存在干扰作用,所以其准确性可以得到有效保证,也可以反映样品中淀粉添加量的真实情况。且实验中采用超纯水涡旋洗涤肉松样品,可以比乙醇水溶液更有效除去可溶性糖类。
葡萄糖在紫外检测其中没有吸收,故不能直接采用紫外检测器 [33-34] 。本实验对葡萄糖进行衍生化处理,使之与PMP反应,该衍生试剂具有强紫外吸收性,且因产物在反相色谱柱中可以保留,在紫外波长245 nm处有强吸收以及无立体异构的特性可建立葡萄糖柱前衍生的方法 [35-36] 。
配制葡萄糖质量浓度为5~50 mg/L系列标准溶液,以质量浓度-峰面积计算线性回归方程,Y=54 053.4X-53 945.9,相关系数r为0.999 8,以仪器响应与基线噪音比(R SN =3)计算,仪器检出限为0.4 μg/mL,方法检出限为0.16%。色谱图见图1。
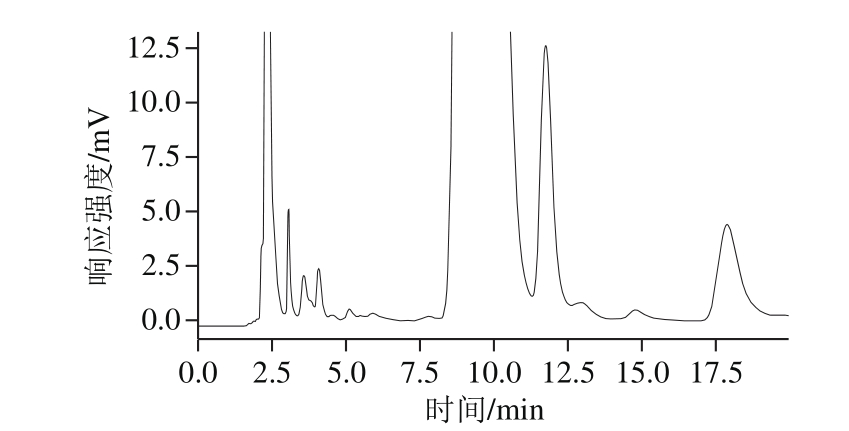
图1 葡萄糖PMP衍生物高效液相色谱图
Fig. 1 Chromatogram of glucose PMP derivative
表3 加标回收率实验结果(n=6)
Table 3 Recoveries of spiked meat floss samples (n= 6)
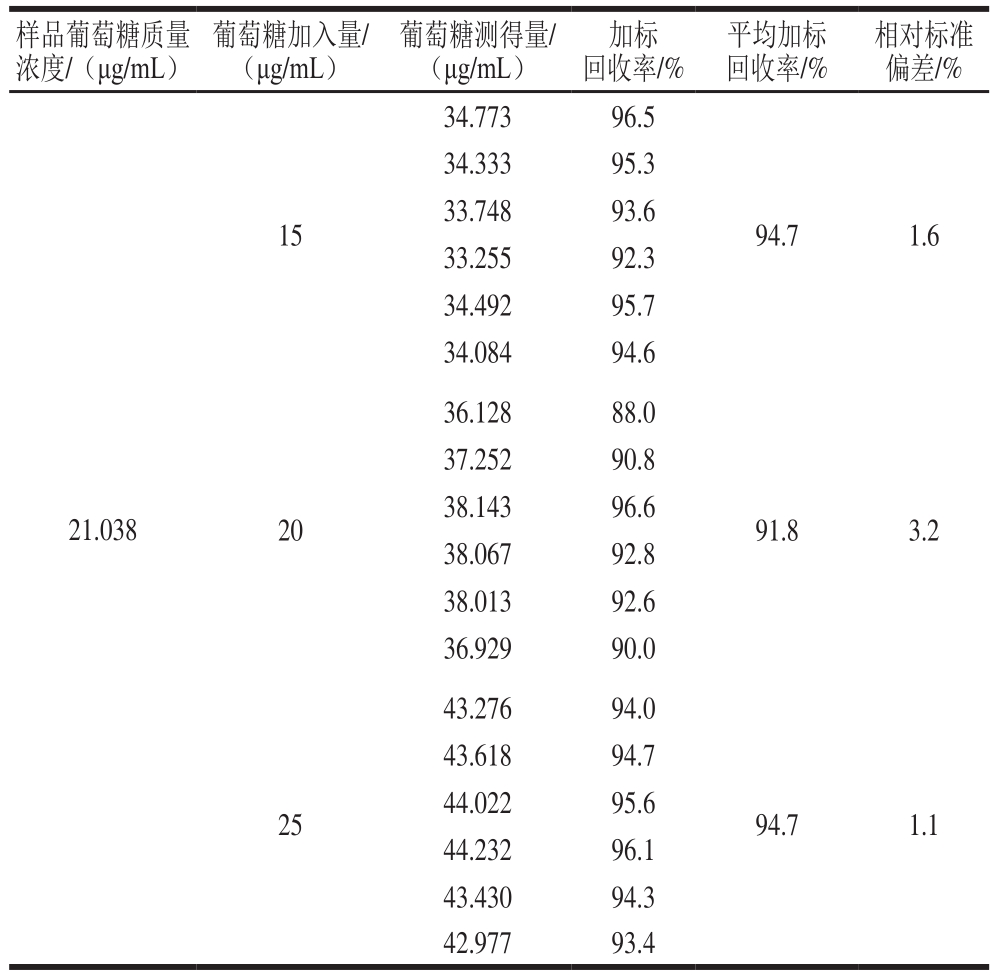
精密称取已知质量浓度葡萄糖肉松水解液6 份,加入已知质量浓度的葡萄糖标准品适量,进行3 个水平加标测定回收率,每个水平进行6 个平行测定。如表3所示,样品回收率在88.0%~96.6%之间,方法的相对标准偏差(日内精密度)小于5%(n=6)。说明本方法适用于肉松中葡萄糖的含量测定。
表4 样品重复性实验结果
Table 4 Repeatability of the HPLC method

称取搅拌均匀的1 g肉松6 份,按酶解条件:液化酶加入量1 000 μL,液化温度100 ℃,超声时间20 min,液化时间60 min,糖化酶加入量100 μL,糖化温度65 ℃,糖化时间60 min进行水解,按1.3.2.2节衍生化,按1.3.2.3节色谱条件进行测定,记录峰面积,计算葡萄糖质量浓度。如表4所示,相对标准偏差为4.62%,表明方法重复性良好。
取经衍生化后的葡萄糖对照品溶液,按1.3.2.3节方法连续进行6 次测定,记录峰面积,如表5所示,相对标准偏差为0.37%,以此表明方法的精密度符合要求。
表5 样品精密度实验结果
Table 5 Precision of the HPLC method

表6 肉松样品测定结果
Table 6 Comparison of actual and measured values of starch content in different real samples

利用1.3.2节方法,对市场上购买的肉粉松、肉绒样品进行测定,如表6所示,淀粉质量分数分别为0%、8.6%、9.4%、10.4%、11.6%、12.1%、12.9%、13.4%、18.2%。从而验证了该方法在实际样品检测中具有操作性强、专一性高、检测灵敏度高等特点。
淀粉的准确定性定量分析是开展肉松食品加工与检测的必备技术基础。利用α-淀粉酶和淀粉葡萄糖苷酶水解淀粉是国际公认的最佳酶解方法 [37] ,目前已有一些采用普通理化方法测定肉松中淀粉的研究报道,但采用高效液相色谱进行测定的报道较少 [38-40] 。本实验建立基于超声辅助双酶水解肉松的高效液相色谱法,考察方法的线性范围、线性方程、检出限、加标回收率及相对标准偏差,相关系数r大于0.999,回收率在88.0%~96.6%之间,相对标准偏差小于5%(n=6),检出限为0.16%。不同于国家标准中的方法,本实验可有效排除大豆可溶性多糖类及其他辅料的干扰,实现了方法回收率及灵敏度提高、检测结果定性定量准确、稳定性好、分析时间缩短等目的。该方法的建立,是对现有标准的改进,对政府部门开展相关产品的风险监测和检测部门开展相应的常规分析检测具有重要的技术支持作用。
参考文献:
[1] LIU C, JIANG S, ZHANG S, et al. Characterization of edible corn starch nanocomposite films: the effect of self-assembled starch nanoparticles[J]. Starch-Stärke, 2016, 68(3/4): 239-248. DOI:10.1002/star.201500252.
[2] SVIHUS B, HERVIK A K. Digestion and metabolic fates of starch,and its relation to major nutrition-related health problems: a review[J].Starch-Stärke, 2016, 68(3/4): 302-313. DOI:10.1002/star.201500295.
[3] COPELAND L, BLAZEK J, SALMAN H, et al. Form and functionality of starch[J]. Food Hydrocolloids, 2009, 23(6): 1527-1534. DOI:10.1016/j.foodhyd.2008.09.016.
[4] AUBRUN-FILLÂTRE C, MONCHAU F, HIVART P. Acrylic bone cement and starch: botanical variety impact on curing parameters and degradability[J]. Materials Science and Engineering: C, 2016, 69:1328-1334. DOI:10.1016/j.msec.2016.08.023.
[5] MESHRAM M W, PATIL V V, MHASKE S T, et al. Graft copolymers of starch and its application in textiles[J]. Carbohydrate Polymers,2009, 75(1): 71-78. DOI:10.1016/j.carbpol.2008.06.012.
[6] YAN Z, LIU Q, DENG Y, et al. Improvement of paper strength with starch modified clay[J]. Journal of Applied Polymer Science, 2005,97(1): 44-50. DOI:10.1002/app.21727.
[7] 栾金水. 栾肉制品中淀粉含量的测定及其研究[J]. 肉类研究, 2004,18(2): 31-34. DOI:10.3969/j.issn.1001-8123.2004.02.012.
[8] 钟姣姣. 肉制品加工中的淀粉性能与应用[J]. 科技信息, 2008(34):205. DOI:10.3969/j.issn.1001-9960.2008.34.165.
[9] 王盼盼. 肉制品加工中使用的辅料: 增稠剂[J]. 肉类研究, 2011,25(2): 29-35. DOI:10.7506/rlyj1001-8123-201102008.
[10] WANG S, COPELAND L. Molecular disassembly of starch granules during gelatinization and its effect on starch digestibility: a review[J]. Food & Function, 2013, 4(11): 1564-1580. DOI:10.1039/C3FO60258C.
[11] SKREDE G. Comparison of various types of starch when used in meat sausages[J]. Meat Science, 1989, 25(1): 21-36. DOI:10.1016/0309-1740(89)90063-6.
[12] LABANCA R A, SILVA C M G, GLÓRIA M B A. Starch levels in refrigerated and frozen chicken based meat products[J]. Brazilian Archives of Biology and Technology, 1999, 42(2): 181-186.DOI:10.1590/S1516-89131999000200008.
[13] FUENTES C, ZIELKE C, PRAKASH M, et al. The effect of baking and enzymatic treatment on the structural properties of wheat starch[J]. Food Chemistry, 2016, 213: 768-774. DOI:10.1016/j.foodchem.2016.07.045.
[14] 何孟杭. 双酶水解-高效液相色谱法测定肉松中淀粉含量的研究[J]. 福建分析测试, 2013, 22(5): 59-62. DOI:10.3969/j.issn.1009-8143.2013.05.15.
[15] 国家卫生和计划生育委员会. 食品中淀粉的测定: GB 5009.9—2016[S]. 北京: 中国标准出版社, 2016: 3-12.
[16] 国家质量监督检验检疫总局. 肉制品 淀粉含量测定: GB/T 9695.14—2008[S]. 北京: 中国标准出版社, 2008: 4-7.
[17] 卫生部. 食品中淀粉的测定: GB/T 5009.9—2008[S]. 北京: 中国标准出版社, 2008: 3-7.
[18] 蔡志兴. 肉松制品淀粉含量测定中干扰物质的研究[J]. 福建分析测试, 2013, 22(6): 49-52. DOI:10.3969/j.issn.1009-8143.2013.06.14.
[19] 田艳玲, 孙妍, 张曼玲, 等. 肉制品(广式腊肠)中淀粉含量测定方法的研究[J]. 粮油加工, 2010(12): 177-179.
[20] WANG Q, FANG Y. Analysis of sugars in traditional Chinese drugs[J].Journal of Chromatography B, 2004, 812(1): 309-324. DOI:10.1016/j.jchromb.2004.09.027.
[21] KUANG H, XIA Y, LIANG J, et al. Fast classification and compositional analysis of polysaccharides from TCMs by ultraperformance liquid chromatography coupled with multivariate analysis[J]. Carbohydrate Polymers, 2011, 84(4): 1258-1266.DOI:10.1016/j.carbpol.2011.01.014.
[22] YANG X, ZHAO Y, WANG Q, et al. Analysis of the monosaccharide components in Angelica polysaccharides by high performance liquid chromatography[J]. Analytical Sciences, 2005, 21(10): 1177-1180.DOI:10.2116/analsci.21.1177.
[23] LV Y, YANG X, ZHAO Y, et al. Separation and quantification of component monosaccharides of the tea polysaccharides from Gynostemma pentaphyllum by HPLC with indirect UV detection[J]. Food Chemistry, 2009, 112(3): 742-746. DOI:10.1016/j.foodchem.2008.06.042.
[24] ZHENG Q, REN D, YANG N, et al. Optimization for ultrasoundassisted extraction of polysaccharides with chemical composition and antioxidant activity from the Artemisia sphaerocephala Krasch seeds[J]. International Journal of Biological Macromolecules, 2016,91: 856-866. DOI:10.1016/j.ijbiomac.2016.06.042.
[25] TIAN H, YIN X, ZENG Q, et al. Isolation, structure, and surfactant properties of polysaccharides from Ulva lactuca L. from South China Sea[J]. International Journal of Biological Macromolecules, 2015, 79:577-582. DOI:10.1016/j.ijbiomac.2015.05.031.
[26] CHEN Z, ZHANG W, TANG X, et al. Extraction and characterization of polysaccharides from Semen Cassiae by microwave-assisted aqueous two-phase extraction coupled with spectroscopy and HPLC[J]. Carbohydrate Polymers, 2016, 144: 263-270. DOI:10.1016/j.carbpol.2016.02.063.
[27] DAI J, WU Y, CHEN S, et al. Sugar compositional determination of polysaccharides from Dunaliella salina by modified RP-HPLC method of precolumn derivatization with 1-phenyl-3-methyl-5-pyrazolone[J].Carbohydrate Polymers, 2010, 82(3): 629-635. DOI:10.1016/j.carbpol.2010.05.029.
[28] SUN L J, MENG Y, SUN J, et al. Characterization, antioxidant activities and hepatoprotective effects of polysaccharides from prepressing separation Fuji apple peel[J]. CyTA-Journal of Food, 2017,15(2): 307-319. DOI:10.1080/19476337.2016.1263241.
[29] ZHANG J, ZHANG Q, WANG J, et al. Analysis of the monosaccharide composition of fucoidan by precolumn derivation HPLC[J]. Chinese Journal of Oceanology and Limnology, 2009, 27(3): 578-582.DOI:10.1007/s00343-009-9205-0.
[30] 张璐瑶, 赵峡, 陈欢欢. 糖类化合物PMP衍生分析进展[J]. 分析测试学报, 2016, 35(3): 367-372. DOI:10.3969/j.issn.1004-4957.2016.03.020.
[31] 林雪. 糖类物质的PMP(1-苯基-3-甲基-5-吡唑啉酮)衍生化及HPLC和MALDI-TOF分析[D]. 西安: 西北大学, 2006.
[32] 陈岩. 肉制品中淀粉含量两种标准检测方法的比较[J]. 肉类工业,2016(11): 28-29. DOI:10.3969/j.issn.1008-5467.2016.11.009.
[33] 刘有平, 张佳蕊, 邵华, 等. 柱前衍生化RP-HPLC法测定地黄中葡萄糖, 半乳糖和蜜二糖[J]. 中草药, 2009(6): 974-976. DOI:10.3321/j.issn:0253-2670.2009.06.046.
[34] 周斌, 张承明, 张承聪, 等. 烟草中糖类的1-苯-3-甲基-5-吡唑啉酮衍生化与HPLC分析方法研究[J]. 云南化工, 2006, 33(6): 39-43.DOI:10.3969/j.issn.1004-275X.2006.06.011.
[35] 贾鹏禹, 孙蕊, 李良玉, 等. 柱前衍生化高效液相色谱检测木糖液中葡萄糖含量[J]. 黑龙江八一农垦大学学报, 2012, 24(3): 41-44.DOI:10.3969/j.issn.1002-2090.2012.03.013.
[36] 张海珠, 史志婷, 王莹, 等. 柱前衍生化高效液相色谱分析云南松松塔多糖的单糖组成[J]. 中国实验方剂学杂志, 2015, 21(24): 30-33.DOI:10.13422/j.cnki.syfjx.2015240030.
[37] 张继红. 食品中淀粉的酶: 比色法分析[J]. 食品工业科技, 2004,25(4): 136-138. DOI:10.3969/j.issn.1002-0306.2004.04.046.
[38] 黄会秋. 微波皂化水解法测定肉制品中淀粉[J]. 中国卫生检验杂志,2008, 18(1): 81-82. DOI:10.3969/j.issn.1004-8685.2008.01.035.
[39] 孙英鸿, 杨晓波, 齐懿鸣. 肉制品中淀粉含量测定方法的比较及关键点[J]. 生命科学仪器, 2013, 11(5): 32-35. DOI:10.3969/j.issn.1671-7929.2013.05.005.
[40] 孙莉. 肉制品中淀粉含量测定方法的比较及关键点[J]. 粮食流通技术, 2016(10): 1-2. DOI:10.16736/j.cnki.cn41-1434/ts.2016.10.001.
Determination of Starch Content in Meat Floss by High Performance Liquid Chromatography
WENG Chenhui 1,2 , CHEN Xiaozhen 1,2, *, LIN Weixing 3 , WU Hong 4 , JIN Zhiyu 3
(1. College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China; 2. Zhejiang Institute of Product Quality and Safety Inspection, Hangzhou 310013, China; 3. Zhejiang DNAZ Testing Technology Co. Ltd.,Hangzhou 310024, China; 4. Hangzhou Weixin Food Co. Ltd., Hangzhou 310052, China)
Abstract: A high performance liquid chromatographic (HPLC) with ultrasonic-assisted two-step enzymatic pretreatment for the determination of the starch content in meat products was developed. After pretreatment, the sample was separated on a C 18 column (4.6 mm × 250 mm, 5 μm), using acetonitrile-20 mmol/L ammonium acetate (22:78, V/V) as mobile phase at a flow rate of 1.0 mL/min, and detected by an ultraviolet detector at a wavelength of 245 nm. The calibration curve was linear in the range of 5–50 mg/L with a correlation coeきcient (r) of 0.999. The average recoveries of spiked hydrolysate were between 88.0% and 96.6% with relative standard deviations (RSDs) of less than 5% (n = 6). The limit of detection (LOD)was 0.4 μg/mL, and the limit of quantitation (LOQ) of the method was 0.16% by using α-amylase and amyloglucosidase.With the advantages of high repeatability and recovery over the national standard methods, this method is suitable for the determination of starch in meat products.
Keywords: high performance liquid chromatography; hydrolysis; meat floss; starch; detection
WENG Chenhui, CHEN Xiaozhen, LIN Weixing, et al. Determination of starch content in meat floss by high performance liquid chromatography[J]. Food Science, 2018, 39(18): 280-285. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201818043. http://www.spkx.net.cn
翁晨辉, 陈小珍, 林卫星, 等. 高效液相色谱法测定肉松中的淀粉含量[J]. 食品科学, 2018, 39(18): 280-285. DOI:10.7506/spkx1002-6630-201818043. http://www.spkx.net.cn
文章编号: 1002-6630(2018)18-0280-06
引文格式:
中图分类号: TS237
文献标志码: A
*通信作者简介: 陈小珍(1960—),女,教授级高级工程师,本科,研究方向为食品安全与检测。E-mail:cxz730@163.com
DOI: 10.7506/spkx1002-6630-201818043
基金项目: 国家标准计划项目(20142110-T-469)
第一作者简介: 翁晨辉(1992—),女,硕士研究生,研究方向为食品安全与检测。E-mail:498949570@qq.com
收稿日期: 2017-07-09