表1 样品信息
Table 1 Information about the microbial samples
玛依乐·艾海提,西热娜依·阿布力克木,努尔古丽·热合曼*
(新疆师范大学生命科学学院,特殊环境物种保护与调控生物学实验室,新疆 乌鲁木齐 830054)
摘 要:采用高通量焦磷酸测序技术和传统培养法对3 份阿图什和1 份乌什传统发酵酸奶进行微生物群落多样性分析。结果表明:4 份样品在3 种培养基上的微生物数量和种类存在差异,即从MRS、YGC及 Lee氏培养基上得到的质量控制后细菌有效序列为20 669条,真菌质量控制后有效序列为293 677 条。在细菌水平上,厚壁菌门(Firmicutes)为各样品中占优势细菌门,丰富度最高,其次为变形菌门(Proteobacteria);属于厚壁菌门的乳杆菌属(Lactobacillus)和链球菌属(Streptococcus)为其中占优势细菌属。在真菌水平上,子囊菌门(Ascomycota)为占优势真菌门,其次为担子菌门(Basidiomycota);属于子囊菌门的酵母菌属(Saccharomyces)和克鲁维酵母属(Kluyveromyces)为占优势真菌属。本研究结果为新疆传统发酵酸奶微生物资源开发、应用提供了微生物多样性依据。
关键词:传统发酵酸奶;高通量测序;微生物多样性
新疆维吾尔自治区维吾尔族长久以来保持饮食发酵酸奶的好习惯,传统发酵酸奶是最常见的自制乳制品之一,富含多种必须的维生素、蛋白质、碳水化合物、脂肪等。传统发酵酸奶具有独特并稳定的微生物群落,具有地区差异性,长期以来所形成的习惯、气候的不同等原因引起了酸奶微生物种类的差异。
高通量测序技术是以一次并行对几十万到几百万条DNA分子进行序列测定为标志。高通量454焦磷酸测序技术为微生物生态领域提供了更准确地识别微生物类群的信息,包括难培养微生物[1-2];对于不同的研究对象该测序技术不仅为提供了大量数据,而且全面证实了研究对象所含微生物的多样性[3]。近几年来高通量测序技术在乳制品行业里广泛应用,彻底克服了传统培养技术及分子鉴定方法的不足,为乳制品微生物群落组成提供更准确和详细的理解,并在优良微生物资源开发和食品安全方面作出了巨大的贡献[4-5],虽然高通量焦磷酸测序法无法识别样品中活和死的微生物,但是该方法有一定的应用前途[6]。第2代测序技术最具有代表性的是Roche公司的454焦磷酸测序技术、Illumina公司的Solexa技术和ABI公司的SOLiD技术,454焦磷酸测序技术目前应用最广泛[7-8]。
前人在实验室条件下培养微生物,并利用分子鉴定方法对新疆部分地区传统酸奶进行了多样性研究,传统研究微生物生态学的方法基本上都局限于用固体培养基分离微生物[9]。然而,随着鉴定技术的发展,更多的研究表明,每一培养基都具有选择性,因此在一定程度上低估了生物环境中微生物种类,而且从固体培养基分离出来的菌落再一次降低了微生物被鉴定的可能性。
本研究采用高通量454焦磷酸测序技术对来自于新疆阿图什及乌什的传统发酵酸奶中微生物类群进行了分析,探讨新疆部分地区酸奶中微生物多样性,从而弥补了微生物在挑菌落不全面、没有真正显示出微生物类群的漏洞。本研究不仅为保护我国传统乳制品中优良菌株资源,而且能为今后传统发酵乳制品的工业化生产提供一定理论依据。
2016年12月分别来自于居住在新疆阿图什、乌什等地区的农民家庭采集的4 份样品:1号样品来自于阿图什阿湖乡光明村544号,3号样品来自于阿图什上阿图什乡拉依力克村吐喀其拉路2号,4号样品来自于阿图什上阿图什拉依力克村阿图什路514号,7号样品来自于阿克苏乌什县阿克托海乡阿克托海村3组23号家庭。采样时利用无菌吸管把样品吸取并立刻装入50 mL的无菌离心管中,用封口膜紧密地包装并立即装在冰袋中立刻运输到实验室,进行后续实验[10]。
胶回收试剂盒 上海生工生物公司;聚合酶链式反应(polymerase chain reaction,PCR)用酶 宝生物工程(大连)有限公司。
电泳槽、离心机、冰箱、LRH-250生化培养箱 上海一恒科技有限公司;光学显微镜 宁波舜宇仪器有限公司;高压蒸汽灭菌锅 上海博讯实业有限公司医疗设备厂;恒温干燥箱 上海天呈实验仪器制造有限公司。
1.3.1 微生物的培养
用0.85%的无菌生理盐水将样品按稀释涂布法进行梯度稀释并将10-5、10-6、10-7稀释度的稀释液分别涂布于改良MRS、Lee氏等培养基(37 ℃)、YGC琼脂平板(28 ℃)培养48 h[11]。最后用无菌水冲洗平板并保存,进行后续实验[12]。平板冲洗液编号及对应的培养基具体信息见表1。
表1 样品信息
Table 1 Information about the microbial samples
1.3.2 总DNA提取
分别取4 份样品平板洗液2 mL置于离心管中振摇确保样品充分混合,12 000 r/min离心5 min弃去上清液,悬浮于1 mL磷酸盐缓冲液(pH 8),12 000 r/min离心10 min,该过程重复3 次[13]。细胞颗粒上加567 µL TE缓冲液和100 µL溶菌酶后悬浮,并在37 ℃加热1 h。再加入60 µL十二烷基硫酸钠(sodium dodecyl sulfate,SDS)溶液(10%)、10 µL蛋白酶K,置于37 ℃水浴2 h,之后添加50 µL NaCl和80 µL十六烷基三甲基溴化铵(cetyltrimethylammonium ammonium bromide,CTAB)-NaCl,在65 ℃加热30 min。沉淀加入等体积的酚-氯仿-异戊醇(25∶24∶1,V/V)溶液12 000 r/min离心10 min(离心2 次收集上清液)。上清液转移到新的离心管,加入等体积氯仿-异戊醇(24∶1,V/V)溶液,轻轻混合12 000 r/min离心5 min,然后把上清液转移到一个新的离心管中,加入0.1体积的3 mol/L NaAc溶液和2 倍体积的冰冷异丙醇,置于-20 ℃孵育30 min。在12 000 r/min离心2 min后将DNA沉淀,用500 µL 70%乙醇溶液洗涤DNA,并自然干燥[14]。最终纯化的DNA溶于50µL TE缓冲液进一步应用。
1.3.3 PCR扩增细菌16S rDNA V4区和真菌ITS区
以稀释D N A为模板,根据测序区域的选择,使用带B a r c o d e引物和高效高保真的酶进行P C R[15]。细菌1 6 S r D N A V 4区引物为5 1 5 F(5’-GTGCCAGCMGCCGCGGTAA-3’)和909R(5’-CCCCGYCAA-TTCMTTTRAGT-3’)。PCR扩增条件:94 ℃、3 min;94 ℃、40 s,56 ℃、60 s,72 ℃、60 s,共循环30 次;72 ℃、5 min。PCR扩增体系:基因组模板DNA 2 μL,2×Easy TaqTMPCR SuperMix 12.5 μL,引物27F 0.5 μL,引物1495R 0.5 μL,ddH2O补足至25 μL。
真菌ITS扩增区引物为ITS4(5’-TCCTCCGCTTATTGATATGC-3’)和gITS7F(5’-GTGART- CATCGART CTTTG-3’)。PCR扩增条件:94 ℃、5 min;94 ℃、30 s,56 ℃、30 s,68 ℃、45 s,共循环34 次;72 ℃、10 min[16]。PCR扩增体系:引物NL1 1.5 μL,引物NL2 1.5 μL,Super Mix 12.5 μL,基因组模板DNA 2.0 μL,ddH2O补足至50 μL。
根据PCR产物浓度进行等量混样,充分混匀后使用2%的琼脂糖凝胶电泳检测对目的条带使用上海生工公司提供的胶回收试剂盒回收产物,并用Nanodrop进行浓度和质量的测定。
1.3.4 文库构建和上机测序
使用TruSeq® DNA PCR-Free Sample Preparation Kit建库试剂盒进行文库构建,构建好的文库经过Qubit和实时荧光定量PCR,文库合格后,使用v2测序试剂盒(2×250 bp)和MiSeq测序仪进行上机测序[16]。
常见的Alpha多样性指数有Shannon多样性指数、Chao1丰富度指数、Observed species(即某个测序深度下观察到的操作分类单元(operational taxonomic units,OTU)数目、Simpson指数等。本研究中所得到的Alpha多样性指数及序列信息参见表2。通过测序,拼接得到的细菌原始序列为211 965 条,质量控制后的有效序列为206 696 条;拼接得到的真菌原始序列为305 252 条,质量控制后的有效序列为293 677 条。该序列根据97%相似性度水平进行OTU划分及嵌合体检查等方面需要深度分析。
表2 3 种培养基上可培养微生物Alpha多样性指数
Table 2 Alpha diversity values of culturable microbes from naturally fermented milk on the three media
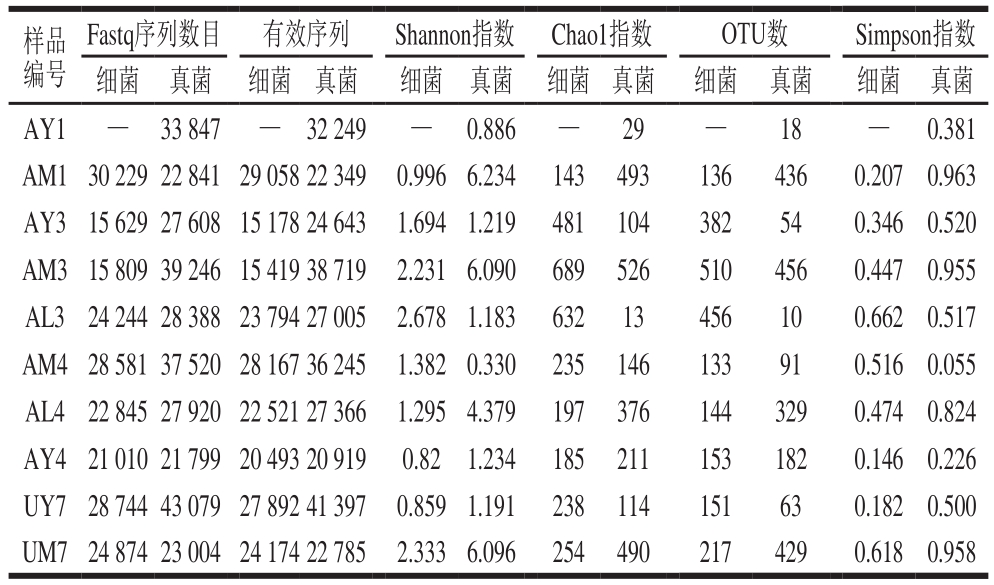
注:—.样品中未发现细菌。
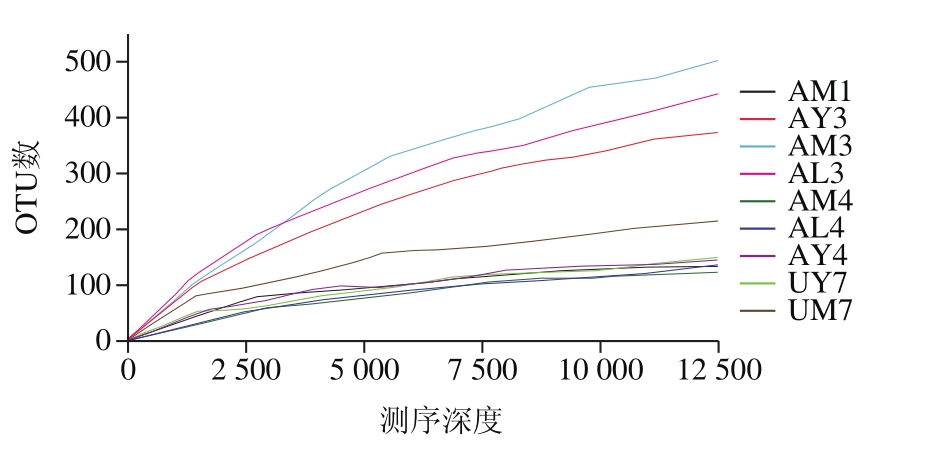
图1 细菌稀释性曲线
Fig. 1 Rarefaction curves of bacteria

图2 真菌稀释性曲线
Fig. 2 Rarefaction curves of fungi
稀疏曲线可根据比较测序数据量不同的样品中物种的丰富度,说明测序数据量是否合理、测序深度是否可靠地表现出微生物组成[17]。稀释曲线趋于平坦说明测序深度合理以及更多的测序量会产生新的OTU;从图1可以看出,样品AM3、AL3、AY3的稀释曲线在小于12 000序列的条件下均没有进入平台期,假如继续增加测序量,OTU数量也会随着增加。其他样品(AM1、AM4、AL4、AY4、UY7、UM7)的测序量大于7 500序列时基本满足样品中细菌丰富度。
由图2可以得出,AM3、AY3、UY7、AM1样品稀释曲线没有趋于平台期,随测序深度的增加也可以出现新的OTU,其他几个样品(AY1、AL3、AM4、AL4、AY4)的稀释曲线到5 000序列为止已进入平缓状态,此结果表明测序深度已足够反映出样品中真菌含量。
PCoA是将聚类分析与主成分分析方法结合起来,用较少的主坐标对分类单元进行有效地排序。在16S rRNA基因序列分析中,普遍使用基于Unifrac距离的PCoA分析,若样本组成越相似反映在PCoA图中的距离越近[18]。UniFrac是通过计算序列在系统发育树上的进化距离,进而比较不同微生物群落间结构差异的一种系统发育进化方法[19]。

图3 细菌基于加权Unifrac距离的PCoA图
Fig. 3 PCoA of bacterial diversity
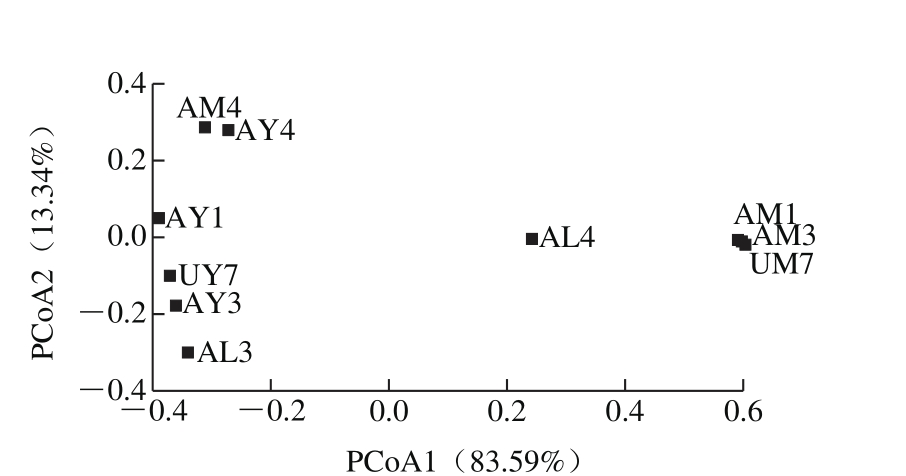
图4 真菌基于加权Unifrac距离的PCoA图
Fig. 4 PCoA of fungi diversity
由图3可知,4 份样品在3 种培养基(MRS、YGC、Lee 氏培养基)上细菌丰富度均独立,没有出现重叠现象,可说明在3 种培养基上4 份样品细菌群落结构具有显著的差异。从图4可知,AM1、AM3、UM7相互重叠,可说明1、3、7号样品在MRS培养基上的真菌群落相似。AY1、AY3、UY7、AY4没出现重叠,可说明1、3、4、7号样品在YGC培养基上真菌群落组成相互独立。而AL3、AL4完全分离。
2.4.1 细菌群落结构分析
为探索被测样品物种多样性信息,对所有样品的有效序列进行聚类,以97%的序列相似性将序列聚类成为OTU再深度分析[20]。如表3所示,在16S rRNA门水平上九个样本共检测到15 个菌门,厚壁菌门是绝对优势菌门,占据94.6%~99.92%,其次是变形菌门占据0.028%~9.38%。1号样品(AM1)相对含量由大到小为99.38%厚壁菌门、0.175 8%变形菌门,其他门丰度小于0.1%。3号样品在MRS及Lee氏培养基上(AM3、AL3)厚壁菌门相对含量均为 99.86%,YGC培养基上(AY3)占优势的为厚壁菌门90.49%,其次为变形菌门9.38%,其他门相对含量小于0.02%。4号样品MRS及Lee氏培养基上(AM4、AL4)厚壁菌门相对含量分别为99.92%、99.51%,除变形菌门外其他门相对含量小于0.1%;YGC培养基上(AY4)占有97.26%的厚壁菌门、1.88%的变形菌门和0.007%的Euryarchaeota,其他门相对含量小于0.77%。7号样品MRS培养基(UM7)中厚壁菌门为99.82%,其他门相对含量小于0.08%;YGC培养基(UY7)有占94.6%的厚壁菌门、4.881%变形菌门,其他门相对含量小于0.04%。由以上数据可看出,4 份样品在3 种培养基上厚壁菌门为绝对优势菌门,相对含量最高,而AY3里的变形菌门含量相比其他样品较多。在AY4中发现含量较多的放线菌门。
表3 在门水平上样品中细菌相对含量(序列所占比例)
Table 3 Relative abundance of bacteria in each sample at phylum level%


图5 细菌累积图(属水平)
Fig. 5 Relative abundance of bacteria at genus level
9 个样品中共测出40 个菌属,其中乳酸菌及链球菌为优势菌(图5)。1号样品在MRS培养基(AM1)上乳杆菌属(Lactobacillus)为绝对优势菌。3号样品在MRS培养基上(AM3)优势菌属为乳杆菌属(92.99%),其次为占6.685%的链球菌属(Streptococcus);Lee氏培养基上(AL3)优势菌属为链球菌(54.96%)属,其次为乳杆菌属(39.18%);YGC培养基上(AY3)除占90.22%的乳杆菌属外,还有占9.262%的醋酸杆菌属(Acetobacter)。4号样品AM4、AL4及AY4里未鉴定属均占优势。7号样品在MRS培养基上(UM7)优势菌属为链球菌属(66.32%),其次为乳杆菌属;YGC培养基上UY7大多数为未鉴定均属,还有少量的醋酸杆菌(7.789%)。
2.4.2 真菌群落结构分析
表4 在门的水平上样品中真菌相对含量(序列所占比例)
Table 4 Relative abundance of fungi in each sample at phylum level%

基于ITS测序门水平,10 个样本中共检测到6 个门,具体数据见表4。1号样品在MRS培养基(AM1)真菌组成成分中以子囊菌门、担子菌门、接合菌门为主,相对含量分别为65.68%、18.87%、10.23%;YGC培养基上(AY1)子囊菌门(99.98%)为绝对优势门。3号样品在MRS培养基上(AM3)真菌以子囊菌门、担子菌门、接合菌门为主,相对含量分别为72.67、17.08、5.404%;Lee氏培养基(AL3)上真菌主要是子囊菌门(99.97%);YGC培养基上(AY3)有99.69%子囊菌门和少量的担子菌门(0.216%)。4号样品在MRS、YGC、Lee氏培养基(AM4、AY4、AL4)上子囊菌门是绝对优势真菌,相对含量分别为99.43%、79.7%、98.21%;Lee氏培养基上还有13.74%担子菌门、3.536%接合菌门。7号样品MRS培养基上(UM7)以66.33%子囊菌门,19.59%担子菌门,8.879%接合菌门为主;YGC培养基上(UY7)以99.74%子囊菌门和少量的担子菌门为主。由以上数据可看出子囊菌门为优势菌门。
基于ITS序列分析在3 种培养基上真菌属水平的组成具体信息参图6。1号样品在MRS培养基(AM1)蜡壳耳属(Sebacina)较多,其次为被孢霉属(Mortierella)、曲霉菌属(Aspergillus)、念珠菌属(Candida)、链格孢属(Alternaria)等;YGC培养基上(AY1)优势菌属为酵母菌属(Saccharomyces)、克鲁维酵母属(Kluyveromyces),相对含量分别为75.35%、24.26%。3号样品在MRS培养基(AM3)真菌里蜡壳耳属较多,其次为被孢霉属、曲霉菌属、念珠菌属、链格孢属;YGC培养基上(AY3)优势菌酵母菌属为52.75%,其次为克鲁维酵母属45.65%;Lee氏培养基上(AL3)里克鲁维酵母属占5.95%,其次为41.93%酵母菌属。4号样品MRS培养基(AM4)酵母菌属为绝对优势菌属占97.19%,还有微量的Mycocentrospora属;Lee氏培养基上(AL4)里酵母菌属较多,其次为蜡壳耳属和被孢霉属、曲霉菌属等。AY4里酵母菌属为优势菌属占87.92%,其次为念珠菌属。7号样品MRS培养基(UM7)里蜡壳耳属和被孢霉属较多,还有少量的曲霉菌属、念珠菌属等;YGC培养基上(UY7)酵母菌属和克鲁维酵母属优势菌分别占60.06%和38.26%。
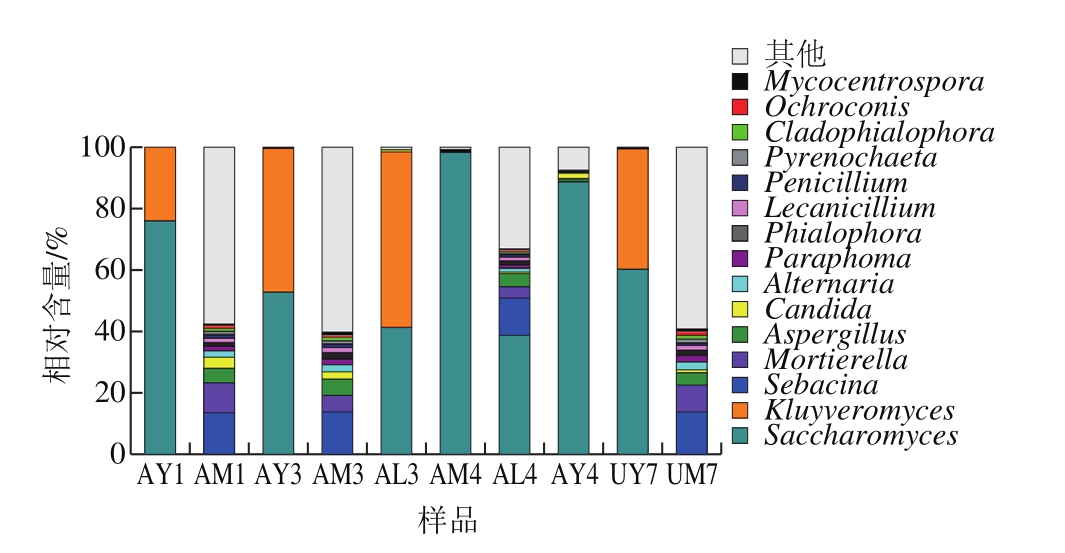
图6 真菌累积图(属水平)
Fig. 6 Relative abundance of fungi at genus level
本研究采用高通量焦磷酸测序技术对3 份阿图什和1 份乌什传统发酵酸奶(MRS、YGC及Lee氏固体培养基上)中的微生物类群进行分析,结果发现:在细菌水平上,4 份样品中厚壁菌门和变形菌门占绝对优势,其中乳杆菌属、链球菌属、醋酸杆菌属、乳球菌属、肠球菌属、芽孢杆菌属的含量较多;在真菌水平上,样品中子囊菌门和但子菌门占绝对优势,其中阿图什酸奶中酵母菌属的含量比乌什酸奶高,但是乌什酸奶中克鲁维酵母属的含量比阿图什酸奶较高。综上所述,厚壁菌门和子囊菌门在各样品中占绝对优势,担子菌门、接合菌门和变形菌门等在每一个样品中都存在,但含量较少。基于MRS和Lee氏培养基上微生物的种类及其含量有差异,在MRS培养基上低丰富度的乳杆菌属占优势,在Lee氏培养基上低丰富度的链球菌属占优势,在YGC培养基上酵母菌属比克鲁维酵母属占优势。
诸多学者对乳制品中的微生物多样性进行研究,基于传统培养方法,鉴定出新疆酸奶中乳杆菌属、肠球菌属、链球菌属、乳球菌属和明串珠菌属[21-23];利用MRS培养基对新疆不同地区酸奶中的优势乳杆菌属进行分离鉴定[24-26]。智楠楠等[27]利用高通量测序技术,对从市场购买的9种酸奶样品进行微生物多样性研究,测定结果表明:酸奶中优势菌为厚壁菌门,这与本实验结果具有一定共同性的同时也存在一定的差异。
本研究结果中微生物多样性特征与其他学者研究结果大体相同,但微生物种类及其数量上具有差异。两个地区的酸奶样品中微生物类群在数量、种类等方面具有一定的差异,这可能是以下几个原因而引起的:采样地区的地理位置、气候、农民制作酸奶的方法、制作工艺(如接种量、接种温度、发酵时间,发酵温度、使用的容器等)、饮食习惯等客观因素的不同引起酸奶浓度及其成分的不同,因此不同地区酸奶中的微生物多样性具有一定的差异[28]。
本实验中存在未测定微生物可能是因为宏基因组测序数据中包含着未知数量的多个物种,所以很难确定一段DNA序列来源于何种物种,拼接时也难以判断何种DNA序列片段应该拼接到一起[29]。本课题组仅研究了4 份样品在3 种培养基上的微生物多样性,对所培养出的微生物组成进行分析,虽然有一定的局限性,但是也能在一定程度上反映出4 份传统发酵酸奶中微生物群落。
参考文献:
[1] SOGIN M L, MORRISON H G, HUBER J A, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”[J]. Proceedings of the National Academy of Sciences of the United States of America,2006, 103(32): 12115-12120. DOI:10.1073/pnas.0605127103.
[2] 米其利, 李雪梅, 管莹, 等. 高通量测序在食品微生物生态学研究中的应用[J]. 食品科学, 2016, 37(23): 302-308. DOI:10.7506/spkx1002-6630-201623049.
[3] 李桥, 王龙龙. 454高通量测序技术在土壤微生物中的应用[J]. 绿色科技, 2013(8): 203-205. DOI:10.3969/j.issn.1674-9944.2013.08.092.
[4] 张敏, 张艳, 兰国伟, 等. 高通量测序技术在乳制品微生物多样性中的研究进展[J]. 中国农学通报, 2016, 32(32): 48-52. DOI:10.11924/j.issn.1000-6850.casb16040016.
[5] QUIGLEY L, OSULLIVAN O, BEWASFORD T P, et al. Highthroughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses[J]. Applied &Environmental Microbiology, 2012, 78(16): 5717-5723. DOI:10.1128/AEM.00918-12.
[6] LIU W, XI X, SUDU Q, et al. High-throughput sequencing reveals microbial community diversity of Tibetan naturally fermented yak milk[J]. Annals of Microbiology, 2015, 65(3): 1741-1751.DOI:10.1007/s13213-014-1013-x.
[7] SHENDURE J J I H. Next-generation DNA sequencing[J]. Nat Biotechnol, 2008, 26: 1135-1145. DOI:10.1038/nbt1486.
[8] 秦楠, 栗东芳, 杨瑞馥. 高通量测序技术及其在微生物学研究中的应用[J]. 微生物学报, 2011, 51(4): 445-457. DOI:10.13343/j.cnki.wsxb.2011.04.013.
[9] 谷峻, 石成芳, 吴晓磊, 等. 油藏微生物群落研究的方法学[J]. 生态学报, 2007(1): 323-328. DOI:10.3321/j.issn:1000-0933.2007.01.038.
[10] 西热娜依·阿布力克木, 穆耶赛尔·玉苏普, 努尔古丽·热合曼. 南疆传统发酵酸奶中可产生生物膜乳酸菌的筛选及鉴定[J]. 食品与发酵工业, 2017, 43(7): 128-133. DOI:10.13995/j.cnki.11-1802/ts.013974.
[11] 谢丽斯. 发酵食品中乳酸菌分离鉴定及生物被膜介导其耐药性的研究[D]. 广州: 广东工业大学, 2012: 12-14.
[12] 呼斯楞, 刘红新, 于洁, 等. 内蒙古呼伦贝尔地区传统发酵乳中乳酸菌的多样性分析[J]. 微生物学通报, 2016(5): 984-990.DOI:10.13344/j.microbiol.china.150919.
[13] 杨洋, 张伟, 袁耀武, 等. 用于PCR检测的乳品中金黄色葡萄球菌DNA提取方法比较研究[J]. 食品与发酵工业, 2005(9): 95-99.DOI:10.13995/j.cnki.11-1802/ts.2005.09.026.
[14] 张申, 王杰, 高江原. 分子生物学检验技术[M]. 武汉: 华中科技大学出版社, 2013.
[15] 肖礼. 黄土丘陵区梯田土壤微生物群落和活性特征及其影响因素[D].杨凌: 西北农林科技大学, 2017: 15-17.
[16] 徐占成, 徐姿静, 刘孟华, 等. 高通量测序法对剑南春大曲真菌群落结构的分析[J]. 酿酒科技, 2018(4): 22-25; 32. DOI:10.13746/j.njkj.2017330.
[17] 王有昭, 朱彤, 谢元华, 等. 基于高通量测序技术的阳极生物膜群落结构[J]. 东北大学学报(自然科学版), 2016, 37(1): 105-108.DOI:10.12068/j.issn.1005-3026.2016.01.022.
[18] 华蔚颖. 应用454测序技术分析菌群结构的方法学研究[D]. 上海:上海交通大学, 2010: 8-9.
[19] 周中凯, 杨雪. 分析包子在不同加工与储藏条件下的菌群多样性[J]. 粮食与油脂, 2017, 30(9): 39-42. DOI:10.3969/j.issn.1008-9578.2017.09.011.
[20] 徐雪雪. 基于高通量测序的马铃薯沟垄覆膜连作土壤微生物多样性分析[D]. 兰州: 甘肃农业大学, 2016: 10-14.
[21] 赵蕊, 霍贵成. 新疆酸奶子中乳酸菌多样性分析[J]. 山东大学学报(理学版), 2008, 43(7): 18-22.
[22] 德亮亮, 乌仁图雅, 秦艳婷, 等. 分离传统酸马奶中乳酸菌的最适培养基的筛选[J]. 中国乳品工业, 2014, 42(8): 4-7. DOI:10.3969/j.issn.1001-2230.2014.08.001.
[23] 古丽奴尔·吐拉西, 普燕, 张富春. 哈萨克族酸奶疙瘩中两种乳杆菌的分离与鉴定[J]. 中国乳品工业, 2014, 42(1): 24-26. DOI:10.3969/j.issn.1001-2230.2014.01.007.
[24] 董晓婉, 李宝坤, 李开雄, 等. 新疆蒙古族和哈萨克族传统乳制品中乳酸菌多样性的比较[J]. 食品工业科技, 2013, 34(21): 162-166.DOI:10.13386/j.issn1002-0306.2013.21.059.
[25] 李远, 巴吐尔, 张小燕, 等. 新疆哈萨克族传统发酵驼乳中乳酸菌的分离鉴定[J]. 中国食物与营养, 2011, 17(1): 54-58. DOI:10.3969/j.issn.1006-9577.2011.01.014.
[26] 秦艳婷. 新疆地区传统发酵乳制品中乳酸菌的分离鉴定及生物多样性分析[D]. 呼和浩特: 内蒙古农业大学, 2014: 9-11.
[27] 智楠楠, 宗凯, 杨捷琳, 等. Illumina Miseq平台深度测定酸奶中微生物多样性[J]. 食品工业科技, 2016, 37(24): 78-82. DOI:10.13386/j.issn1002-0306.2016.24.007.
[28] 张敏, 张艳, 黄丽丽, 等. 基于16S rDNA高通量测序方法比较新疆西北部地区乳品中微生物的多样性[J]. 食品科学, 2017, 38(20): 27-33.DOI:10.7506/spkx1002-6630-201720005.
[29] 李俊锋. 基于16S rRNA和宏基因组高通量测序的微生物多样性研究[D]. 北京: 清华大学, 2015: 19-21.
Diversity of Culturable Microorganisms in Traditional Fermented Milk from South Xinjiang as Analyzed by High-Throughput Pyrosequencing
Mayira AHAT, Xirinay ABLIKIM, Nurgul RAHMAN*
(Laboratory of Special Species Conservation and Regulatory Biology, School of Life Sciences,Xinjiang Normal University, Ürümqi 830054, China)
Abstract:High-throughput pyrosequencing and the traditional culture method were used in this study to analyze the microbial community diversity in four samples of traditional fermented milk collected from south Xinjiang. Results showed that there were differences in the number and species of microbes isolated from the four samples when they were cultured on three media, i.e., MRS, YGC and Lee media. A total of 20 669 effective bacterial and 293 677 effective fungal sequences were obtained after quality control. Firmicutes was the dominant bacterial phylum in all samples with the highest abundance followed by Proteobacteri; Lactobacillus and Streptococcus were found to the dominant genera in the Firmicutes phylum.Ascomycota was the dominant fungal phylum followed by Basidiomycota; the genera Saccharomyces and Kluyveromyces were the dominant fungi belonging to the Ascomycota phylum. The results of this study can provide the basis for the exploitation and utilization of microbial resources in traditional fermented milk in Xinjiang.
Keywords:fermented milk; high-throughput pyrosequencing; microbial diversity
Mayira AHAT, Xirinay ABLIKIM, Nurgul RAHMAN. Diversity of culturable microorganisms in traditional fermented milk from south Xinjiang as analyzed by high-throughput pyrosequencing[J]. Food Science, 2018, 39(20): 126-131. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201820019. http://www.spkx.net.cn
引文格式:玛依乐·艾海提,西热娜依·阿布力克木,努尔古丽·热合曼. 应用高通量测序法检测南疆传统酸奶中微生物多样性[J].食品科学, 2018, 39(20): 126-131. DOI:10.7506/spkx1002-6630-201820019. http://www.spkx.net.cn
文章编号:1002-6630(2018)20-0126-06
文献标志码:A
中图分类号:TS201.3
DOI:10.7506/spkx1002-6630-201820019
*通信作者简介:努尔古丽•热合曼(1972—),女,副教授,博士,研究方向为应用微生物。E-mail:nurgulum@163.com
第一作者简介:玛依乐•艾海提(1985—),女,硕士研究生,研究方向为应用微生物。E-mail:277829574@qq.com
基金项目:国家自然科学基金地区科学基金项目(31460448)
收稿日期:2017-06-20