王向阳,顾 双,杨 玲
(浙江工商大学食品与生物工程学院,浙江 杭州 310018)
摘 要:用橘霉素人工抗原-佐剂免疫牛蛙,研究牛蛙肌肉蛋白质对橘霉素结合能力的大小。将能与橘霉素结合的蛋白质用海藻酸钠制粒。取6.4 mg橘霉素人工抗原免疫牛蛙肌肉蛋白质,使用海藻酸钠质量浓度4 g/100 mL、氯化钙质量浓度5 g/100 mL、戊二醛交联体积分数0.2%、交联时间2 h,制备海藻酸钙包埋蛋白质凝胶微球。取海藻酸钙凝胶微球2 g装填成固相小柱,以2 mL红曲米橘霉素提取液过柱,以20 mL超纯水洗脱杂质,与市售免疫亲和柱色谱图比较,均没有杂质干扰峰。以橘霉素标准液作为上样液,以70%乙腈-三氟乙酸作为橘霉素洗脱液,洗脱剂量为5 mL,回收率为86.7%,柱容量为0.101 µg/g。采用自制的牛蛙免疫蛋白柱和市售免疫亲和柱测得红曲米中橘霉素含量分别为0.037 µg/g和0.043 µg/g。
关键词:橘霉素;牛蛙;免疫亲和柱
橘霉素是真菌产生的一种次级代谢产物,主要由红曲霉、青霉和部分曲霉产生[1]。橘霉素对肾脏具有强烈的毒性[2],还具有细胞毒性[3-4],能增加人体过敏反应[5],具有致畸性和胚胎毒性[6],还能损害肝的代谢,有致癌性,可诱发突变等[7]。1991年,橘霉素被国际生命科学院自然毒素检测委员会欧洲分会列为必须检测的毒素之一;2012年3月欧洲食品安全局食物链污染物科学专家组认为橘霉素是肾毒素,且橘霉素在果汁和蔬菜汁[8]、小麦[9]、红曲米[10]和酿酒用曲[11]中均有发现。目前从青霉素中发现2 个橘霉素衍生物pencitrin和pencitrinol,其基团和橘霉素差异较大,另外还有橘霉素的二聚体[12]。
高效液相色谱(h i g h p e r f o r m a n c e l i q u i d chromatography,HPLC)法是目前检测橘霉素最普遍的方法,另外还有高液相色谱-质谱联用[13]、超高液相色谱-串联质谱[14-15]和荧光比色法[16]。免疫亲和层析常与高效液相色谱-荧光检测器(high performance liquid chromatography with fluorescence detector,HPLC-FLD)联用。GB/T 5009.222—2008《红曲类产品中桔青霉素的测定》和SN/T 2916—2011《出口食品中桔霉素的测定方法 免疫亲和柱净化-高效液相色谱法》橘霉素的测定,都是采用免疫亲和柱对样品进行去杂纯化,再进行HPLCFLD检测[17-18]。美国维康公司等提供橘霉素免疫亲和柱,价格昂贵、保存条件苛刻,且保质期短,其制作方法未见报道。橘霉素能进入环状糊精内部空间,增强稳定性[19],如果进行较长时间浸泡,可能会吸附较多橘霉素,未来或可作为橘霉素的一种富集纯化的手段。
陈福生等[20]用蛋清白蛋白-橘霉素连接物免疫小白鼠,制备单克隆抗体,建立酶联免疫吸附实验(enzymelinked immunosorbent assay,ELISA)法检测红曲样品橘霉素含量。Duan Zhihui等[21]合成橘霉素人工抗原,免疫母鸡,从鸡蛋中提取和纯化得到抗体,建立ELISA法检测橘霉素。Abramson等[22]将橘霉素与匙孔血蓝蛋白偶联制备人工抗原免疫兔子,获得多克隆抗体,建立ELISA测定大麦中的橘霉素。汪媛媛等[23]利用双交联法制备橘霉素-蛋白质偶联抗原免疫小鼠获得多克隆抗体,建立ELISA检测橘霉素。Kononenko等[24]制得葡萄糖氧化酶-橘霉素偶联抗原和牛血清白蛋白-橘霉素抗原,得到兔抗血清,建立检测橘霉素的ELISA方法。
前人大多研究用橘霉素人工抗原免疫小鼠、兔子等获得抗体,但用牛蛙制备抗体用于免疫检测或免疫亲和实验还鲜见报道。本研究前期实验发现,橘霉素直接注射牛蛙肌肉蛋白对橘霉素的吸附量比对照仅提高5%~15%,而使用橘霉素人工抗原免疫牛蛙后,其肌肉蛋白对橘霉素的吸附量比对照提高100%以上。说明其具有很好的吸附作用,且牛蛙肌肉蛋白量远比血清多,也容易获得。海藻酸钠常被用作固定化酶的载体[25],其与钙离子形成凝胶微球,具有三维网状结构,被称为“egg-box”结构[26],固定化可减少混入的蛋白酶对牛蛙肌肉蛋白的分解。本研究将牛蛙肌肉蛋白用海藻酸钠包埋制粒后制柱,可用于样品中橘霉素检测的纯化预处理,然后与HPLC-FLD相结合,用于食品中橘霉素的检测,其成本远比市售免疫亲和柱低。目前橘霉素标准品很贵,化学脱除橘霉素的方法对食品品质影响较大,如果能够使用牛蛙肌肉蛋白吸附橘霉素,未来可以用于标准品制备,以及液体食品中橘霉素的快速脱除。
红曲米和牛蛙购于杭州市下沙物美超市。
海藻酸钠 杭州禾德化工有限公司;免疫亲和柱、橘霉素标准品 北京泰乐祺科技有限公司;弗氏完全佐剂 杭州邦易化工有限公司。
LC 20-A HPLC仪、RF-20A荧光检测器 日本岛津公司;高速冷冻离心机 北京五洲东方科技有限公司。
1.3.1 橘霉素人工抗原免疫牛蛙肌肉蛋白制备
免疫用人工抗原的制备[27]:50 µg橘霉素溶解于500 µL甲醇中,加入1 mL 0.1 mol/L NaHCO3溶液,再加入40 µL 37%的甲醛溶液,混匀后,滴入1 mL 5 mg/mL牛血清白蛋白(bovine serum albumin,BSA)溶液中,37 ℃振摇反应24 h后,移入透析袋中,置0.01 mol/L磷酸缓冲液(phosphatic buffer solution,PBS)4 ℃透析,每3 h换一次透析液,透析12 h,透析完成后冷冻干燥。再将干燥后的橘霉素-BSA人工抗原用0.65%生理盐水溶解,使橘霉素-BSA质量浓度为10 µg/mL,取0.5 mL橘霉素-BSA溶液与1.5 mL弗氏完全佐剂混合乳化。
免疫实验[28]:前期实验发现用橘霉素人工抗原注射牛蛙后,饲养16 d时,牛蛙大腿的可溶性蛋白质含量最高,该蛋白对橘霉素的吸附量也最高。因此,采用大腿肌肉注射,注射2.5 µg/mL的橘霉素人工抗原-佐剂,注射剂量为每只牛蛙0.1 mL,并养殖16 d。
肌肉蛋白提取:取牛蛙大腿肌肉1.0 g,剪碎,放入50 mL离心管中,加入10 mL PBS(0.01 mol/L pH 7.4,含1 mmol/L EDTA、1 mmol/L焦磷酸钠、1% Tween-20),超声破碎,10 000 r/min离心20 min后取上清液,即为肌肉组织总蛋白提取液,4 ℃冰箱密封保存。蛋白质含量采用考马斯亮蓝比色法测定。
1.3.2 处理牛蛙肌肉蛋白与橘霉素的结合能力测定
将2.5 µg/mL橘霉素人工抗原-佐剂和2.5 µg/mL橘霉素分别注射牛蛙,注射剂量为每只牛蛙0.1 mL,饲养16 d和24 d,提取牛蛙肌肉蛋白。之后将肌肉蛋白提取液1 mL放于10 mL玻璃管中,加入1 mL 1.5 µg/mL的橘霉素标准品,旋涡振荡器混匀,4 ℃条件下3 h,液体装入透析袋,放于50 mL离心管中,加入20 mL 0.01 mol/L pH 7.4的PBS作为透析液,放于4 ℃透析12 h,取透析袋外液体1 mL,HPLC-FLD法测定橘霉素含量,计算如式(1)所示:
式中:Y为肌肉蛋白结合橘霉素量/(µg/mg);m为添加的橘霉素质量/µg;c为透析液中橘霉素质量浓度/(µg/mL);V为透析液体积/mL;n为添加的蛋白质量/mg。
1.3.3 牛蛙肌肉蛋白与海藻酸钠制备微球工艺
0.40 g海藻酸钠加入10 mL 0.64 mg/mL牛蛙肌肉蛋白溶液,磁力搅拌器混合均匀,用5 mL注射器(滴头为医用7号针头)将混合好的溶液逐滴滴入100 mL质量浓度3 g/100 mL的氯化钙溶液中,4 ℃固定2 h,用100 mL蒸馏水洗涤。测定3 g/100 mL氯化钙溶液中的蛋白质量浓度,计算蛋白质的包埋率见式(2)。并随机取出10 粒微球拍照,用Image-Pro Plus 6.0软件分析颗粒的大小。
式中:C1为包埋反应前蛋白质量浓度/(mg/mL);V1为包埋反应前蛋白体积/mL;C2为固定化后氯化钙溶液中蛋白质量浓度/(mg/mL);V2为固定化后氯化钙溶液体积/mL。
1.3.4 微球制备工艺优化
1.3.4.1 海藻酸钠质量浓度的选择
分别取海藻酸钠加牛蛙肌肉蛋白溶液混匀,质量浓度1、2、3、4 g/100 mL,滴入100 mL质量浓度3 g/100 mL的氯化钙溶液中,测定包埋率和颗粒大小。
1.3.4.2 氯化钙质量浓度的选择
0.40 g海藻酸钠4 份,加10 mL牛蛙肌肉蛋白溶液混匀,滴入100 mL质量浓度1、3、5、7 g/100 mL的氯化钙溶液中,测定包埋率和颗粒大小。
1.3.4.3 戊二醛体积分数对免疫球蛋白活性的影响
0.40 g海藻酸钠5 份,加10 mL牛蛙肌肉蛋白溶液混匀,滴入100 mL质量浓度3 g/100 mL氯化钙溶液中。分别加入30 mL体积分数为0.1%、0.2%、0.4%、0.8%、1.0%的戊二醛溶液,4 ℃交联反应4 h,再用100 mL蒸馏水洗涤微球。将洗涤后的2 g微球装入固相小柱,取0.1 µg/mL的橘霉素标准液2 mL,过柱后收集液体,2 mL定容,HPLC-FLD检测橘霉素。
1.3.4.4 交联时间对免疫球蛋白活性的影响
0.40 g海藻酸钠5 份,加10 mL牛蛙肌肉蛋白溶液混匀,滴入100 mL质量浓度3 g/100 mL氯化钙溶液中。用体积分数0.2%的戊二醛在4 ℃交联反应0.5、1、2、3、4 h,再用100 mL蒸馏水洗涤微球。将洗涤后的2 g微球装入固相小柱,取0.1 µg/mL的橘霉素标准液2 mL,过柱后收集液体,2 mL定容,HPLC-FLD检测橘霉素。
1.3.5 橘霉素测定
使用岛津HPLC仪,岛津ODS柱(Hypersil ODS2,250 mm×4.6 mm,5 μm);流动相为乙腈(色谱纯)-三氟乙酸(pH 2.5)体积比60∶40;流速0.8 mL/min;进样量40 µL;柱温28 ℃;检测器为RF-20A荧光检测器,检测波长:λex=331 nm,λem=500 nm。将橘霉素标准液依次稀释,使其质量浓度为1.6、0.8、0.4、0.2、0.1、0.01 µg/mL,制作标准曲线。橘霉素吸附率计算如式(3)所示:
式中:C1为过柱前橘霉素标准液质量浓度/(µg/mL);C2为过柱后橘霉素标准液质量浓度/(µg/mL);V为橘霉素标准液体积/mL。
1.3.6 吸附柱及上样液的制备
在经上述条件优化后制备的海藻酸钙凝胶微球中加入30 mL体积分数0.2%的戊二醛溶液,4 ℃处理2 h。用100 mL蒸馏水洗涤,于0.05 mol/L pH 7.4的Tris-HCl缓冲液中保存。
微球小柱的制备:微球2.0 g装于5 mL注射器,底部用脱脂棉封口后装填,均匀压实,用10 mL 0.05 mol/L p H 7.4的T r i s-H C l缓冲液平衡小柱后,再加入3 mL 0.05 mol/L pH 7.4的Tris-HCl缓冲液作为保护液。
上样液准备:红曲米10.0 g,加50 mL 70%甲醇溶液,超声提取20 min,搅拌10 min,过滤,收集滤液。取5 mL滤液,用醋酸盐缓冲液(0.05 mol/L,pH 6.0)50 mL定容,混匀后玻璃纤维滤纸过滤。
1.3.7 使用条件的优化
1.3.7.1 洗杂液的选择
上样液2 mL加入小柱,作用30 min,去底盖,加压去液体,用20 mL超纯水、20 mL PBS(0.01 mol/L,pH 7.4)作为洗杂液对未被结合的橘霉素和杂质进行洗脱,洗脱完成后,用5 mL 70%乙腈-三氟乙酸溶液作为洗脱液洗脱橘霉素,HPLC-FLD检测。
1.3.7.2 洗脱液的选择
上样标准液的配制:取1 µg/mL的橘霉素标准溶液1 mL,用醋酸盐缓冲液(0.05 mol/L,pH 6.0)定容至20 mL。上样标准液2 mL加入小柱,底盖封口,作用30 min ,去底盖,加压去液体,加20 mL超纯水洗杂,用5 mL 50%~90%甲醇-三氟乙酸溶液和50%~70%乙腈-三氟乙酸溶液作为洗脱液洗脱橘霉素,收集液体,定容至5 mL,HPLC-FLD检测,按式(4)计算橘霉素回收率:
式中:X1为上样标准液中橘霉素质量浓度/(µg/mL);V1为上样标准液的体积/mL;X2为洗脱液中橘霉素质量浓度/(µg/mL);V2为洗脱液的体积/mL。
1.3.7.3 洗脱液用量的确定
取上样标准液1 mL上样,连续加入6 mL 70%乙腈-三氟乙酸作为洗脱液,每1 mL收集一管,HPLC-FLD法测橘霉素含量,计算回收率。
1.3.8 柱容量的测定
取橘霉素质量浓度为0.05 µg/mL上样标准液连续进样5 mL,每1 mL收集一管,HPLC-FLD测橘霉素含量。柱容量计算如式(5)所示:
式中:M1为上样液中橘霉素质量/μg;M2为过柱吸附后橘霉素质量/μg;m为凝胶微球质量/g。
1.3.9 回收率的比较
上述红曲米提取液2 mL,加0.04、0.08、0.16 µg橘霉素标准品后过柱。另取2 mL未添加橘霉素标准品滤液过柱。回收率计算如式(6)所示:
式中:X1为样品未加标时橘霉素质量/µg;X2为样品加标后的橘霉素质量/µg;m为橘霉素标准品质量/µg。
1.3.10 方法平行性精密度实验及检测限测定
取红曲米4 份,每份10 g。前处理按照1.3.6节和1.3.7节中优化的牛蛙肌肉蛋白柱纯化方法,将红曲米样品经过制备好的牛蛙肌肉蛋白柱纯化后,采用HPLC-FLD检测橘霉素,对所得的4次峰面积进行统计分析。取初始质量浓度为0.2 µg/mL橘霉素标准溶液,依次做2 倍比稀释并对各质量浓度进行HPLC-FLD检测分析,取信噪比为3时的质量浓度作为其检测限。
表1 注射橘霉素及其人工抗原牛蛙肌肉蛋白结合橘霉素的能力
Table 1 Citrinin binding ability of bullfrog muscle protein after injection of citrinin-BSA conjugate and citrinin

注:采用t检验,*.差异显著,P<0.05。
如表1所示,在16 d和24 d,橘霉素人工抗原诱导的牛蛙肌肉蛋白结合橘霉素量分别比橘霉素直接诱导增加102.1%和117.1%,说明橘霉素人工抗原诱导牛蛙后,其肌肉中产生了亲和橘霉素的蛋白。
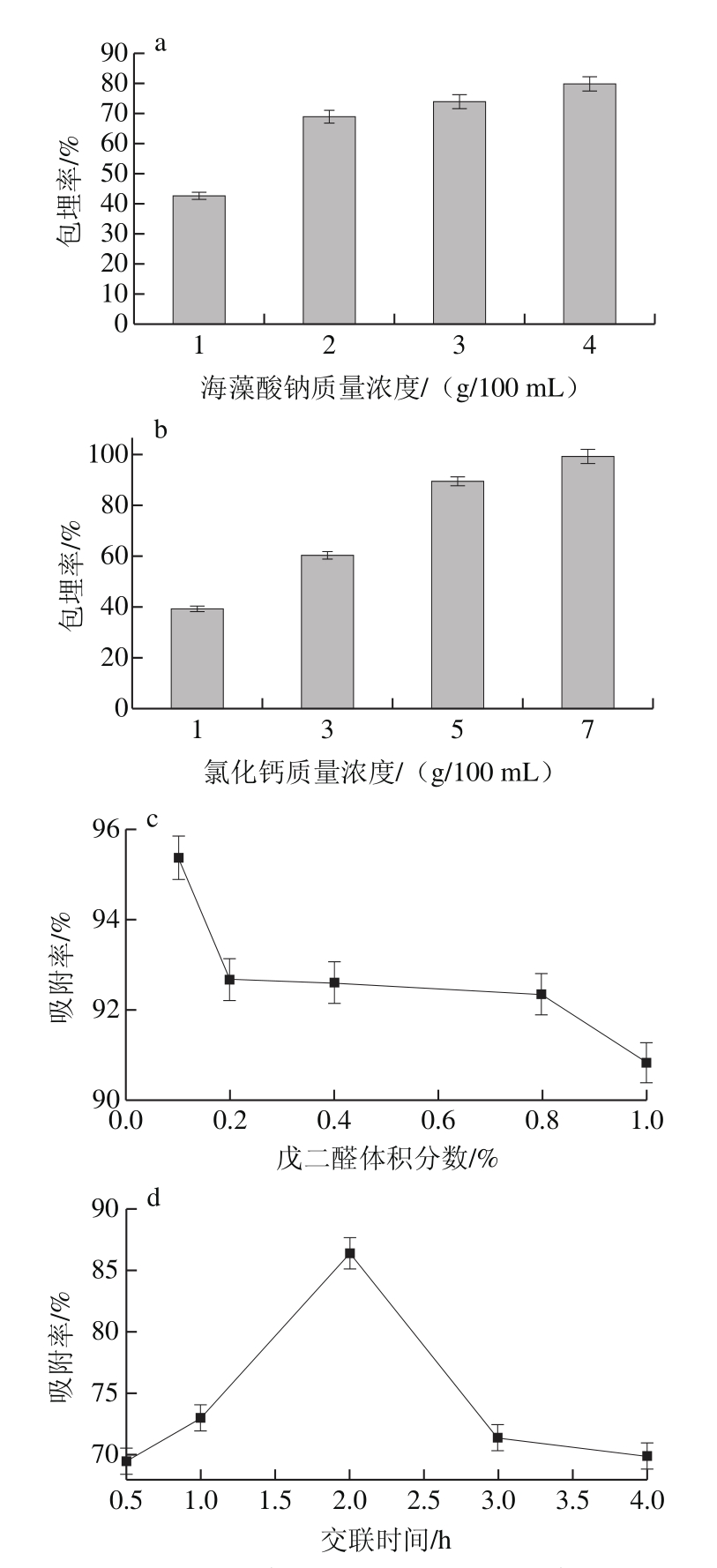
图1 海藻酸钠质量浓度(a)、氯化钙质量浓度(b)、戊二醛体积分数(c)、交联时间(d)分别对蛋白包埋率和活性的影响
Fig. 1 Effects of sodium alginate concentration (a), calcium chloride concentration (b) and glutaraldehyde concentration (c), and crosslinking time (d) on embedding efficiency and citrinin binding capacity
海藻酸钠质量浓度1 g/100 mL不能形成球状,2~4 g/100 mL均能形成直径为2.32~2.57 mm的球状颗粒。随着海藻酸钠质量浓度增加,凝胶微球的直径随着增大,对蛋白质的包埋率也随之升高。质量浓度大于4 g/100 mL时,黏稠度太高,手工滴制困难,因此选择4 g/100 mL的海藻酸钠,其对蛋白质的包埋率为79.6%(图1a)。
1~7 g/100 mL的氯化钙均能制备海藻酸钙凝胶微球,直径为2.29~2.47 mm。随着氯化钙质量浓度的增加,微球直径逐渐减小,但对蛋白质的包埋率增大。形成“egg-box”结构的速度也随之增大,能够使蛋白质更迅速地被包埋在凝胶微球中,提高包埋率,胀大效应减小,从而使微球直径减小[29]。当氯化钙质量浓度为7 g/100 mL时,已成型的凝胶微球浮于氯化钙溶液表面,难以继续滴制,因此选择5 g/100 mL的氯化钙,对蛋白质包埋率可达到89.5%(图1b)。
随着戊二醛体积分数从0.1%增加到1.0%,微球对橘霉素的吸附率降低。使用体积分数为0.1%的戊二醛交联时,洗涤液在280 nm波长处有吸收峰,说明蛋白质结合不牢固,在洗涤过程中被洗脱下来。选择体积分数0.2%的戊二醛作为交联剂,对橘霉素的吸附率为92.7%(图1c)。
包埋后的凝胶微球与体积分数0.2%的戊二醛交联0.5~4 h,在交联时间为0.5~2 h时,微球对橘霉素的吸附率逐渐增大,2 h时达到最大值86.4%,随后继续延长与戊二醛的交联时间,微球对橘霉素的吸附能力反而降低,最适交联时间为2 h(图1d)。

图2 3 种不同柱的柱容量比较
Fig. 2 Comparison of the column capacities of three different columns
a.超纯水洗杂自制柱;b.市售免疫亲和柱;c. PBS洗杂自制柱。
如图2所示,红曲米样品的橘霉素提取液过自制蛋白柱,用超纯水洗杂效果良好,用PBS洗杂导致橘霉素峰信号被掩盖。马萍等[30]研究表明海藻酸钙凝胶微球在酸性介质和蒸馏水中均不溶胀,在pH 6.8的PBS中,很快就会发生溶胀,微球结构被破坏,所以导致无法准确检测橘霉素的色谱峰。而市售免疫亲和柱的材料不同,用PBS作为杂质洗脱液,效果良好。橘霉素提取液经市售免疫亲和柱净化,与超纯水洗杂后经自制柱净化的两者橘霉素出峰保留时间分别为6.264 min和6.297 min,两者基本一致,测得红曲米样品橘霉素的含量分别为0.043 µg/g和0.037 µg/g,自制柱对橘霉素的吸附效率为进口免疫亲和柱的86.05%。
表2 不同洗脱液洗脱后的橘霉素回收率比较
Table 2 Recoveries of citrinin with different eluents

如表2所示,随着甲醇和乙腈体积分数的增加,橘霉素回收率逐渐增大,但是甲醇体积分数为90%时,橘霉素回收率仅为64.3%,乙腈体积分数为60%时,回收率已经达到80%,故选择70%乙腈-三氟乙酸作为橘霉素洗脱液。
随着洗脱液用量的增大,橘霉素更多的被洗脱下来,回收率升高,当洗脱液洗脱至6 mL时,仅有0.001 µg橘霉素被洗脱下来,因此选择5 mL洗脱液进行洗脱(表3)。
表3 洗脱液用量的选择
Table 3 Selection of optimal eluent volume

在连续上样5 mL时,测得前4 mL过柱吸附后的流出液中基本没有橘霉素,说明上样液中98%以上的橘霉素被吸附,而在5 mL过柱吸附后的流出液中则有92%的橘霉素流出,并没有被吸附,说明此时柱对橘霉素的结合量已达到饱和。柱容量为0.101 µg/g(表4)。
表4 柱容量测定
Table 4 Determination of column capacity
如表5所示,在橘霉素添加量为0.04 µg时,市售免疫亲和柱的回收率较高达到94.61%,而自制牛蛙柱回收率为83.93%;当橘霉素添加量为0.08 µg时,自制牛蛙肌肉蛋白柱回收率为78.7%,仅比市售免疫亲和柱低1.59%;当橘霉素添加量为0.16 µg,自制牛蛙肌肉蛋白柱回收率反而稍高于市售免疫亲和柱。
表5 市售免疫亲和柱与自制牛蛙肌肉蛋白柱的回收率比较
Table 5 Recoveries of immunoaffinity column and the column developed in this study

红曲米样品过自制蛋白柱,HPLC-FLD检测橘霉素,对所得的峰面积进行统计分析。该方法的相对标准偏差(relative standard deviation,RSD)值为1.81%,符合中国药典中关于精密度实验RSD应小于3%的规定,因此,该方法具有良好的精密度。取初始质量浓度为0.2 µg/mL橘霉素标准溶液,依次做2 倍比稀释并对各浓度进HPLC-FLD检测分析,取信噪比为3时的质量浓度作为其检测限,测得橘霉素的检测限为0.004 µg/mL(表6)。
表6 方法精密度实验
Table 6 Precision of the method proposed in this study

将橘霉素人工抗原免疫牛蛙肌肉蛋白质用海藻酸钠进行包埋,制备海藻酸钙凝胶颗粒。经过包埋率测试,最佳条件为橘霉素人工抗原免疫牛蛙肌肉蛋白质6.4 mg,选择质量浓度4 g/100 mL的海藻酸钠、质量浓度5 g/100 mL的氯化钙、体积分数0.2%的戊二醛交联2 h制备海藻酸钙凝胶微球。
用2 g海藻酸钙凝胶微球装填成固相小柱,红曲米样品橘霉素提取液2 mL作为上样液,20 mL超纯水洗脱杂质和未被结合的橘霉素,其纯化效果与免疫亲和柱类似,均没有杂质干扰峰。使用70%的乙腈-三氟乙酸作为洗脱液,洗脱剂用量为5 mL时,橘霉素回收率可达到86.7%。橘霉素加标回收率实验表明,虽然自制牛蛙肌肉蛋白柱总体回收率稍低于市售免疫亲和柱,但在橘霉素添加量为0.04 µg时,回收率仍可达到83.93%,柱容量测定值为0.101 µg/g;取信噪比为3时的质量浓度作为其检测限,测得橘霉素检测限为0.004 µg/mL,方法精密度实验RSD为1.8%,符合《中国药典》中关于精密度实验RSD应小于3%的规定,说明该方法具有良好的精密度。在红曲米的橘霉素含量检测中,采用自制的牛蛙免疫蛋白柱和市售免疫亲和柱测得橘霉素含量分别为0.037 µg/g和0.043 µg/g。未来将完善自制牛蛙免疫蛋白颗粒的制作,进一步提高产品性能。
参考文献:
[1] WONG H C, KOEHLER P E. Production and isolation of an antibiotic from Monascus purpureus and its relationship to pigment production[J]. Journal of Food Science, 1981, 46(2): 589-592.DOI:10.1111/j.1365-2621.1981.tb04917.x.
[2] FRIIS P, HASSELAGER E, KROGH P. Isolation of citrinin and oxalic acid from Penicillium viridicatum Westling and their nephrotoxicity in rats and pigs[J]. Acta Pathologica et Microbiologica Scandinavica,1969, 77(3): 559-660. DOI:10.1111/j.1699-0463.1969.tb04263.x.
[3] LORKOWSKI G, CREPPY E E, BECK G, et al. Inhibitory action of citrinin on cultured hepatoma cells[J]. Food and Cosmetics Toxicology,1980, 18(5): 489-491. DOI:10.1016/0015-6264(80)90162-5.
[4] KROGH P, HASSELAGER E, FRIIS P. Studies on fungal nephrotoxicity. II Isolation of two nephrotoxic compounds from Penicillium viridicatum Westtling: citrinin and oxalic acid[J]. Acta Pathologica Et Microbiologica Scandinavica, 1970, 78(4): 401-413.
[5] WICHMANN G, HERBARTH O, LEHMANN I. The mycotoxins citrinin, gliotoxin, and patulin affect interferon-gamma rather than interleukin-4 production in human blood cells[J]. Environmental Toxicology, 2002, 17(3): 211-218. DOI:10.1002/tox.10050.
[6] HOOD R, HAYES A W, SCAMMELL J G. Effects of prenatal administration of citrinin and viriditoxin to mice[J]. Food and Cosmetics Toxicology, 1976, 14(3): 175-183. DOI:10.1016/S0015-6264(76)80419-1.
[7] AMBROSE A M, DEEDS F. Some toxicological and pharmacological properties of citrinin[J]. Journal of Pharmacology and Experimental Therapeutics, 1946, 88(2): 173-186.
[8] DIETRICH R, SCHMID A, MAIRTLBAUER E. Citrinin in fruit juices[J]. Mycotoxin Research, 2001, 17(2): 156-159. DOI:10.1007/BF03036426.
[9] ZAIED C, ZOUAOUI N, BACHA H, et al. Natural occurrence of citrinin in Tunisian wheat grains[J]. Food Control, 2012, 28(1): 106-109. DOI:10.1016/j.foodcont.2012.04.015.
[10] 许赣荣, 卢晨, 穆晓青, 等. 部分红曲霉菌株产桔霉素的研究[J].食品与生物技术学报, 2000, 19(1): 58-61. DOI:10.3321/j.issn:1673-1689.2000.01.015.
[11] 李凤琴, 许赣荣, 李玉伟, 等. 国产红曲制品中桔青霉素污染水平研究[J]. 卫生研究, 2005, 34(4): 451-454. DOI:10.3969/j.issn.1000-8020.2005.04.022.
[12] HU Y M, ZHANG J N, LIU D, et al. Pencitrin and pencitrinol, two new citrinin derivatives from an endophytic fungus Penicillium citrinum salicorn 46[J]. Phytochemistry Letters, 2017, 22: 229-234.DOI:10.1016/j.phytol.2017.10.003.
[13] TUOMI T, JOHNSSON T, HINTIKKA E L, et al. Detection of af l atoxins (G1-2, B1-2), sterigmatocystin, citrinine and ochratoxin A in samples contaminated by microbes[J]. Analyst, 1999, 126(9): 1545-1550. DOI:10.1039/b103790k.
[14] PAMEL V E, VERBEKEN A, VLAEMYNCK G, et al. Ultrahighperformance liquid chromatographic-tandem mass spectrometric multimycotoxin method for quantitat-ting 26 mycotoxins in maize silage[J]. Agricultural and Food Chemistry, 2011, 59(18): 9747-9755.DOI:10.1021/jf202614h.
[15] HAN Z, REN Y P, ZHU J F, et al. Multianalysis of 35 mycotoxins in traditional Chinese medicines by ultra-high-performance liquid chromatography-tandem mass spectrometry coupled with accelerated solvent extraction[J]. Agricultural and Food Chemistry, 2012, 60(33):8233-8247. DOI:10.1021/jf301928r.
[16] 卢健, 黄晓东, 谢飞飞. 荧光分光光度法测定调味品中的桔霉素含量的研究[J]. 安徽工程大学学报, 2014, 29(4): 20-22. DOI:10.3969/j.issn.2095-0977.2014.04.006.
[17] 中国疾病预防控制中心营养与食品安全所. 红曲类产品中桔青霉素的测定: GB/T 5009.222—2008[S]. 北京: 中国标准出版社, 2008.
[18] 山东出入境检验检疫局. 出口食品中桔霉素的测定方法 免疫亲和柱净化-高效液相色谱法: SN/T 2916—2011[S]. 北京: 中国标准出版社, 2011.
[19] POÓR M, MATISZ G, KUNSÁGI-MÁTÉ S, et al. Fluorescence spectroscopic investigation of the interaction of citrinin with native and chemically modified cyclodextrins[J]. Journal of Luminescence,2016, 172: 23-28. DOI:10.1016/j.jlumin.2015.11.011.
[20] 陈福生, 邢淑婕. 红曲产品中桔霉素含量的ELISA测定[J]. 食品科学, 2004, 25(8): 169-172. DOI:10.3321/j.issn:1002-6630.2004.08.040.
[21] DUAN Z H, LIN Z R, YAO H R, et al. Preparation of artificial antigen and egg yolk-derived immunoglobulin (IgY) of citrinin for enzyme-linked immunosorbent assay[J]. Biomedical and Environmental Sciences, 2009,22(3): 237-243. DOI:10.1016/S0895-3988(09)60051-9.
[22] ABRAMSON D, USLEBER E, MARTLBAUER E. Determination of citrinin in barley by indirect and direct enzyme immunoassay[J].Journal of AOAC International, 1996, 79(6): 1325-1329.
[23] 汪媛媛, 李泳宁, 郭养浩. 双交联法制备桔霉素-蛋白质偶联抗原及抗体[J]. 生物化学与生物物理进展, 2010, 37(3): 337-341.
[24] KONONENKO G, BURKIN A. Immunoenzyme method for the determination of citrinin[J]. Journal of Analytical Chemistry, 2007,62(7): 691-696. DOI:10.1134/S1061934807070155.
[25] 李鸿玉, 历重先, 李祖明. 海藻酸钠固定化果胶酶的研究[J]. 食品科技, 2010, 35(4): 21-24.
[26] ISABELLE B, SERGE P. Molecular basis of Ca2+induced gelation in alginates and pectins: the egg-box model revisited[J].Biomacromolecules, 2001, 2(4): 1089-1096.
[27] 刘仁荣, 余宙, 何庆华, 等. 抗桔青霉素单克隆抗体的研制与鉴定[J]. 卫生研究, 2009, 36(2): 190-193. DOI:10.3969/j.issn.1000-8020.2007.02.016.
[28] 杭小英. 牛蛙链球菌病的病原及疫苗研究[D]. 杭州: 浙江大学,2012: 22-23.
[29] 孙艳玲, 张鹏超, 王妮. 海藻酸钙水凝胶纤维的制备及应用[J]. 纺织科学研究, 2014(4): 108-110.
[30] 马萍, 祝力, 孙淑英, 等. 海藻酸钙凝胶微球的制备和pH依赖性溶胀[J]. 中国海洋药物, 2003, 22(5): 35-37. DOI:10.3969/j.issn.1002-3461.2003.05.010.
Preparation and Adsorption Performance of Immunoaffinity Column Based on Muscle Antibody from Bullfrog (Rana catesbeiana) for Detection of Citrinin
WANG Xiangyang, GU Shuang, YANG Ling
(School of Food Science and Biotechnology, Zhejiang Gongshang University, Hangzhou 310018, China)
Abstract:The citrinin binding capacity of muscle protein from bullfrog (Rana catesbeiana) immunized with citrinin-bovine serum albumin (BAS) conjugate emulsified with Freund’s complete adjuvant was investigated. The protein with high citrinin binding capacity was embedded into sodium alginate microspheres. The optimum conditions were determined as follows:immunogen dosage 6.4 mg, sodium alginate concentration 4 g/100 mL, CaCl2concentration 5 g/100 mL, glutaraldehyde concentration 0.2%, and crosslinking time 2 h. Two milliliter of the crude citrinin extract from red rice was passed through a column packed with 2 g of sodium alginate microspheres followed by washing with 20 mL of ultra-pure water to remove the impurities, and good chromatogram was obtained without interfering peaks compared with commercial immunoaffinity column. The recovery of citrinin standard solution eluted with 5 mL of 70% acetonitrile and trif l uoroacetic acid mixture was 86.7%. The column capacity was 0.101 µg/g. The content of citrinin in red rice was determined to be 0.037 and 0.043 µg/g using the column developed in this study and commercial immunoaffinity column, respectively. However, further study should be done to improve the preparation process for better product performance.
Keywords:citrinin; bullfrog; immunoaffinity chromatography
WANG Xiangyang, GU Shuang, YANG Ling. Preparation and adsorption performance of immunoaffinity column based on muscle antibody from bullfrog (Rana catesbeiana) for detection of citrinin[J]. Food Science, 2018, 39(22): 159-165. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201822025. http://www.spkx.net.cn
引文格式:王向阳, 顾双, 杨玲. 橘霉素免疫牛蛙肌肉蛋白柱的制备及其吸附橘霉素作用分析[J]. 食品科学, 2018, 39(22): 159-165.DOI:10.7506/spkx1002-6630-201822025. http://www.spkx.net.cn
文章编号:1002-6630(2018)22-0159-07
文献标志码:A
中图分类号:TS201.3
DOI:10.7506/spkx1002-6630-201822025
第一作者简介:王向阳(1966—),男,教授,博士,研究方向为农产品保鲜与加工和食品安全。E-mail:wxy200228@sina.com
基金项目:浙江省自然科学基金项目(LY15C200005)
收稿日期:2017-09-14