利用高通量测序技术分析民间面引子中的真菌多样性
刘建利,孙 敏,曹晓虹,张 琇,李靖宇
(北方民族大学生物科学与工程学院,发酵酿造工程生物技术国家民委重点实验室,宁夏特殊生境微生物资源开发与利用重点实验室,宁夏 银川 750021)
摘 要:面引子是传统面食加工中发酵剂,以前对其微生物的研究大多集中于分离培养法,本实验旨在采用新一代高通量测序技术揭示传统面引子中的真菌群落结构特征。从我国不同地区收集18 个民间面引子样品,基于宏基因组采用聚合酶链式反应扩增ITS1区,Illumina MiSeq测序,云平台分析样品中真菌群落。结果表明:18 个样品中发现101 个操作分类单元,属于真菌界4 个门、9 个纲、15 个目、25 个科、38 个属、56 个种;酿酒酵母是大多数样品中的优势菌;酿酒酵母、Fungi sp.、unclassified_k__Fungi这3 个种在所有样品中都存在,而且数量较大;Kazachstania bulder、戴尔凯氏有孢圆酵母、矮小假丝酵母、光滑假丝酵母菌、清酒假丝酵母这5 个种数量较大,但只在单个样品中出现;异常威克汉姆酵母、Alternaria sp.和帚状曲霉数量相对多一些,出现频率较高,在绝大多数样品中都有分布。各样品真菌群落结构相差不大,而且类型和地理分布差异也不明显。
关键词:高通量测序;面引子;真菌群落
面引子,又称为酵子、起子、酵头、面头、老酵面、老面、面肥等,与国外面包制作中的酸面团类似,是我国传统的馒头、包子、饼、烤馕等面食制作中用到的发酵剂,可分为“酵子”型和“老面”型,其使用历史最早可以追溯到汉代[1],曾经一段时间在面食制作中传统面引子被商品酵母发酵剂取代,只有在我国北方的小部分农村家庭仍被使用。近年来,随着人们生活水平的提高和消费习惯转型,采用传统面引子制作的面食具有更加细致的质地、细腻的口感和更为丰富的风味重新受到消费者热捧[2-4]。传统面引子的优势在于其中有多种微生物,能引发的多个生化反应,混合协同发酵产生复杂的风味物质[5-8]。因此,利用现代生物技术阐明传统面引子中的微生物种类及其作用机制成为研究热点[9-12]。以前的研究大多集中于采用分离培养方法对其中的酵母菌和乳酸菌进行研究[13-16],偶尔也有采用聚合酶链式反应-变性梯度凝胶电泳(polymerase chain reaction-denaturing gradient gel electrophoresis,PCR-DGGE)技术的报道[17-20]。
高通量测序是最近发展的新一代测序技术,由于其具有测序数据量大、可同时分析多个样品的优势。近年来,已成为分析食品微生物多样性、评价食品发酵和贮藏过程中微生物群落结构变化、监测食源性病原菌动态特征以及控制食品质量等食品微生物分子生态学研究的首选方法,已成功应用于开菲尔、泡菜、奶酪、酸奶、啤酒、葡萄酒、发酵香肠、酸面包、发酵鱼等食品中微生物群落结构的演替规律的研究[21-24]。陈星星[25]采用高通量测序技术分析了面引子中的细菌菌群结构,但鲜有用高通量测序研究面引子中真菌群落的报道,因此,本实验拟采用高通量测序技术揭示来自不同地区民间传统面引子中真菌菌群落结构,为保护传统面引子和开发工业化面引子提供理论依据。
1 材料与方法
1.1 材料与试剂
本实验的酵子样品从我国北方的7 个省采集,18 个样品分别来自于我国河北省、山东省、河南省、陕西省、山西省、甘肃省和吉林省,基质均为小麦,具体地区与样品情况见表1。
表1 样品信息
Table 1 Information about the samples

编号 样品状态 样品类型 采集地点JT1 颗粒状 酵子 河北邯郸市JT2 颗粒状 酵子 河北承德市JT3 颗粒状 酵子 山东滨州市JT4 大块状 酵子 山东青岛市JT5 稀稠状 老面 山东菏泽市JT6 大块状 酵子 山东济南市JT8 稀稠状 老面 山西临汾市洪洞县JT9 颗粒状 酵子 山西运城市JT10 稀稠状 老面 陕西西安市JT11 稀稠状 老面 陕西榆林市JT12 稀稠状 老面 陕西渭南市JT13 稀稠状 老面 河南周口市JT14 稀稠状 老面 河南周口市淮阳县JT15 颗粒状 酵子 河南商丘市JT16 稀稠状 老面 吉林四平市JT17 稀稠状 老面 甘肃临夏州JT18 稀稠状 老面 甘肃白银市景泰县JT19 颗粒状 酵子 甘肃武威市
Power Fecal®DNA Isolation Kit基因组DNA提取试剂盒美国MoBio公司;TransGen AP221-02 TransStart FastPfu DNA Polymerase 北京全式金生物技术有限公司;AxyPrep DNA凝胶回收试剂盒 美国Axygen公司;TruSeqTMDNA Sample Prep Kit 美国Illumina公司;引物由南京金斯瑞生物科技有限公司合成。
1.2 仪器与设备
Nanodrop2000超微量分光光度计 美国Thermo Scientific公司;GeneAmp®9700型PCR仪 美国ABI公司;QuantiFluor™-ST蓝色荧光定量系统 美国Promega公司。
1.3 方法
1.3.1 DNA抽提
参照基因组DNA提取试剂盒说明提取样品中总基因组DNA,超微量分光光度计检测DNA纯度和浓度;1%琼脂糖凝胶,5 V/cm电压,时间20 min检测DNA完整性。
1.3.2 PCR扩增
20 μL反应体系:5×FastPfu Buffer 4 μL;2.5 mmol/L dNTPs 2 μL;Forward Primer(5 μmol/L)0.8 μL;Reverse Primer(5 μmol/L)0.8 μL;FastPfu Polymerase 0.4 μL;牛血清白蛋白0.2 μL;Template DNA 10 ng;补ddH2O至20 μL。PCR参数:1×(95 ℃、3 min),循环数×(95 ℃、30 s;55 ℃、30 s;72 ℃、45 s),72 ℃、10 min。引物序列见表2。
表2 引物序列
Table 2 Sequences of primers used for PCR

测序类型 引物名称 引物序列 扩增区域 长度真菌ITS ITS1F 5’-CTTGGTCATTTAGAGGAAGTAA-3’ ITS1 300 bp左右ITS2(2043R)[26]5’-GCTGCGTTCTTCATCGATGC-3’
为保证后续数据分析的准确性及可靠性,需要两个条件,首先是尽可能使用低循环数扩增;其次保证每个样品扩增的循环数统一。每个样本3 个重复,将同一样本的PCR产物混合后取3 μL上样2%琼脂糖凝胶电泳检测。使用凝胶回收试剂盒切胶回收PCR产物,Tris-HCl洗脱;2%琼脂糖电泳检测。
1.3.3 荧光定量
参照电泳初步定量结果,将P C R产物用QuantiFluor™-ST蓝色荧光定量系统进行检测定量,之后按照每个样本的测序量要求,进行相应比例的混合。
1.3.4 文库构建与MiSeq测序
由安诺优达基因科技(北京)有限公司测序服务完成。
1.3.5 生物信息学分析
MiSeq测序得到的PE reads首先根据overlap关系进行拼接,对序列质量进行质控和过滤,导入交互式微生物多样性云分析平台(http://www.i-sanger.com/,上海美吉生物医药科技有限公司),区分样本后进行操作分类单元(operational taxonomic units,OTU)聚类分析和物种分类学分析,基于OTU信息可以进行多种多样性指数分析,基于分类学信息,可以在各个分类水平上进行群落结构的统计分析。在上述分析的基础上,可以对多样本的群落组成和系统发育信息进行多元分析和差异显著性检验等一系列深入的统计学和可视化分析。参数设置如下:相似度为97%,按最小样品序列数抽平样本序列;选择比对数据库UNITE(Release 6.0 http://unite.ut.ee/index.php)的真菌数据库[27],置信度阈值为0.7;合并低于0.000 5%的OTU;物种、样本层级聚类方式用average,分组的样本丰度计算用均值;多重检验校正fdr,后检验Tukey;多组比较策略one-against-all。
2 结果与分析
2.1 测序样本数据统计
对18 个面引子样品的真菌ITS1区测序,质控后获得677 306 条序列,序列平均长度为388 bp,按照97%相似度分类,共产生101 个OTU。
2.2 Alpha多样性分析
Alpha多样性指数用于研究环境中微生物的多样性。样品中真菌Alpha多样性指数如表3所示,样品中OTU的数目在30~70之间,OTU数目最低的是样品JT1,只有30 个OTU,最高的是样品JT17、JT15和JT8,OTU数目分别是68、67、66。
表3 样品真菌多样性指数
Table 3 Diversity index of fungi in samples

样品 OTU Shannon指数 Simpson指数 Chao1指数 ACE指数 Shannoneven指数Simpsoneven指数 Coverage JT1 30 0.31 0.92 30.00 31.11 0.09 0.04 1.00 JT2 60 1.01 0.47 62.14 63.44 0.25 0.04 1.00 JT3 43 1.35 0.37 43.00 43.34 0.36 0.06 1.00 JT4 46 0.75 0.65 64.33 126.60 0.20 0.03 1.00 JT5 43 0.31 0.91 48.25 49.69 0.08 0.03 1.00 JT6 51 1.04 0.45 54.50 68.95 0.26 0.04 1.00 JT8 66 1.22 0.43 73.20 73.15 0.29 0.04 1.00 JT9 44 0.54 0.84 53.33 71.38 0.14 0.03 1.00 JT10 58 1.16 0.57 63.00 62.59 0.28 0.03 1.00 JT11 36 0.54 0.80 42.00 0.00 0.15 0.03 1.00 JT12 49 0.82 0.64 52.00 55.02 0.21 0.03 1.00 JT13 37 0.81 0.64 44.00 48.38 0.22 0.04 1.00 JT14 44 2.23 0.23 58.00 118.33 0.59 0.10 1.00 JT15 67 0.69 0.80 73.00 74.86 0.16 0.02 1.00 JT16 51 0.92 0.61 61.50 62.29 0.23 0.03 1.00 JT17 68 1.79 0.34 71.75 70.84 0.42 0.04 1.00 JT18 37 2.23 0.24 40.00 40.50 0.62 0.11 1.00 JT19 42 0.40 0.89 52.50 52.22 0.11 0.03 1.00
Shannon指数和Simpson指数是综合衡量物种多样性的指数,Shannon指数值越高,Simpson指数值越低,物种多样性越丰富,反之物种多样性越少。18个样品中,Shannon指数最高、Simpson指数最低的样品为JT14和JT18,反映出样品JT14和JT18中真菌OTU的多样性较高,但这2个样品的OTU数目却不是最多,Shannon指数最低、Simpson指数最高的样品为JT1、JT5和JT19,反映出这3 个样品中真菌多样性较低,但这3 个样品的OTU数目却不是最少,因此,Shannon指数和Simpson指数的高低和样品中OTU数目并不相关;总体来看,大多数酵子型面引子的Shannon指数较低,Simpson指数较高,样品真菌OTU多样性也较低,而大多数老面型面引子的Shannon指数较高,Simpson指数较低,样品中OTU多样性都较高,推测可能因为酵子型面引子样品呈现较干的块状,存放时间较长,里面的真菌较少,或者干块状造成样品DNA提取得率较低,而老面型面引是湿样品,可能更适合微生物生长并保存。
Chao1指数和ACE指数主要用于衡量样本中的物种种类丰富度。Chao1指数主要利用只含有1条序列的OTU数目(singleton)和只含有2条序列的OTU数目(doubleton)判断群落的物种丰富度,它对单个物种的变化更为敏感,它的数值越大,表示物种种类越多;ACE指数是通过singleton和稀有物种(出现次数≤10次)估算还有多少没被发现的物种,当测序中singleton的比例越大,ACE值越大,样本的真实物种种类越多。Chao1指数最高的样品为JT8、JT15 和JT17,这3 个样品的OTU数目也最多,Chao1指数最低的样品为JT1,其OTU数目也最少,可以看出,与Shannon指数和Simpson指数不同,大多数样品的Chao1指数大小和OTU数目正相关;ACE指数最高的样品为JT4和JT14,ACE指数最低的样品为JT11,其ACE指数为0,样品JT1的ACE指数也较低,和Shannon指数和Simpson指数相似,ACE指数大小和样品中OTU数目也不相关。结果中还出现Chao1指数最高最低的样品和ACE指数最高最低的样品不一致的现象,以及同一样品Chao1指数和ACE指数大小表现不一致的现象,可能是因为样品总OTU中singleton、doubleton以及稀有物种在总OTU中的比例不同造成。
Shannoneven指数和Simpsoneven指数反映样本中物种数量分布的均匀程度。Shannoneven指数最高的样品为JT14和JT18,Shannoneven指数最低的样品为JT1、JT5、JT9和JT19,与其Shannon指数表现一致。但Simpsoneven指数却与其Simpson指数表现并不一致,虽然样品JT14和JT18的Simpsoneven指数最高,但其余样品的Simpsoneven指数均很低。总体看,只有样品JT1的Shannon指数和Simpson指数都显示其真菌多样性较低,2个丰富度指数Chao1指数和ACE指数都显示其真菌丰富度较低,2 个均匀度指数Shannoneven指数和Simpsoneven指数都显示其真菌均匀度也较低,其余样品的多样性综合指数、2 个丰富度指数、2 个均匀度指数表现并不完全一致,说明这些样品中真菌种类和数量分布上有不同。
Coverage是指各样本中低丰度OTU的覆盖情况,其数值越高,则样本中序列被测出的概率越高,而没有被测出的概率越低,所有样品真菌的Coverage值都为1,说明测序已经较高覆盖低丰度OTU,表明本次测序结果代表了样本中真菌的真实情况。
2.3 物种组成
本实验18 个样品中出现的101 个OTU,属于真菌界4 个门、9 个纲、15 个目、25 个科、38 个属、56 个种。
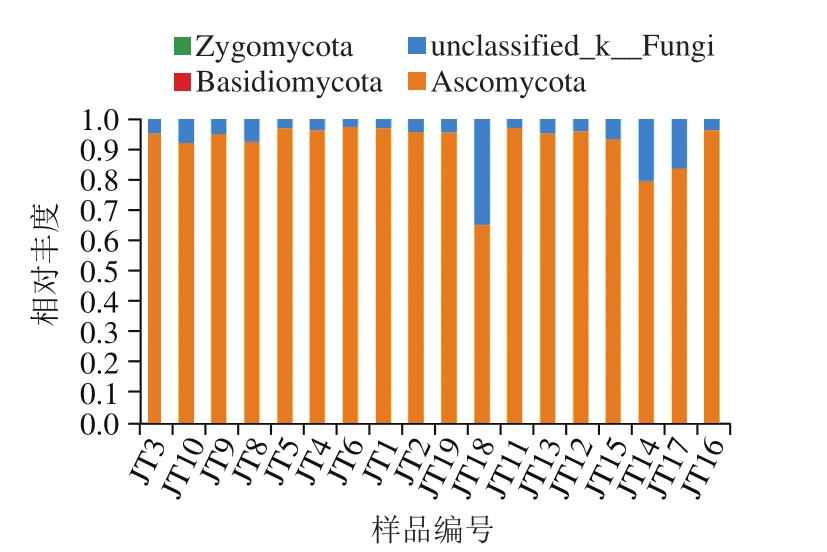
图1 门水平各样品中真菌相对分布柱形图
Fig. 1 Relative abundances of fungi at the phylum level
在样品中门水平分布如图1所示,样品中共出现4 个门,包括3 个已知门:子囊菌门(Ascomycota)、接合菌门(Zygomycota)和担子菌门(Basidiomycota),1 个未鉴定(unclassified_k__Fungi)。所有样品中子囊菌门都占优势,在样品JT6中最高,占97.37%,在样品JT18中最低,占75.1%;unclassified_k__Fungi在所有样品中占丰度第2高;样品JT17中还有极低比例的接合菌门和担子菌门。
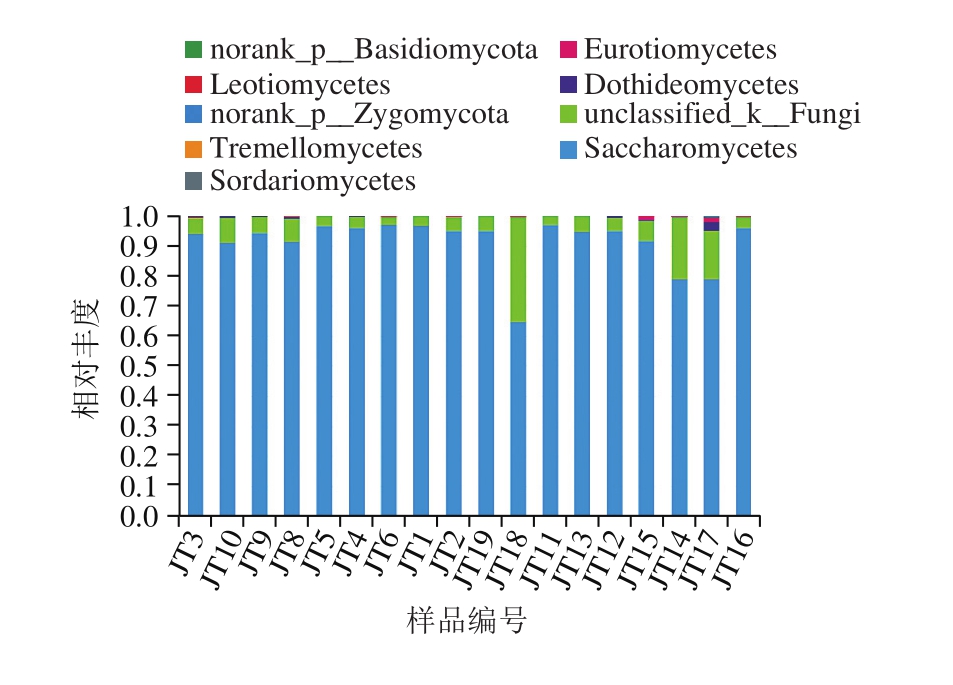
图2 纲水平各样品中真菌相对分布柱形图
Fig. 2 Relative abundance of fungi at the class level
在纲水平分布见图2,样品中共出现的9 个纲,包括6 个已知的纲:酵母菌纲(Saccharomycetes)、座囊菌纲(Dothideomycetes)、散囊菌纲(Eurotiomycetes)、粪壳菌纲(S o r d a r i o m y c e t e s)、银耳纲 (Tremellomycetes)和锤舌菌纲(Leotiomycetes),2个分类不明:分类不明的接合菌(norank_p__Zygomycota)和分类不明的担子菌(norank_p__Basidiomycota),1 个未鉴定出的真菌(unclassified_k__Fungi)。酵母菌纲在所有样品中占优势,unclassified_k__Fungi依然占丰度第2高,样品JT17、JT14、JT10和JT8中丰度第3的是座囊菌纲,样品JT2、JT6、JT15、JT6和JT18中丰度第3的是散囊菌纲,样品JT3、JT9和JT12中丰度第3的是粪壳菌纲;样品JT8和JT12中丰度第4是粪壳菌纲,样品JT15中丰度第4的是座囊菌纲,样品JT17中丰度第4的是散囊菌纲,第5是粪壳菌纲;另外,在一些样品中发现银耳纲和锤舌菌纲。
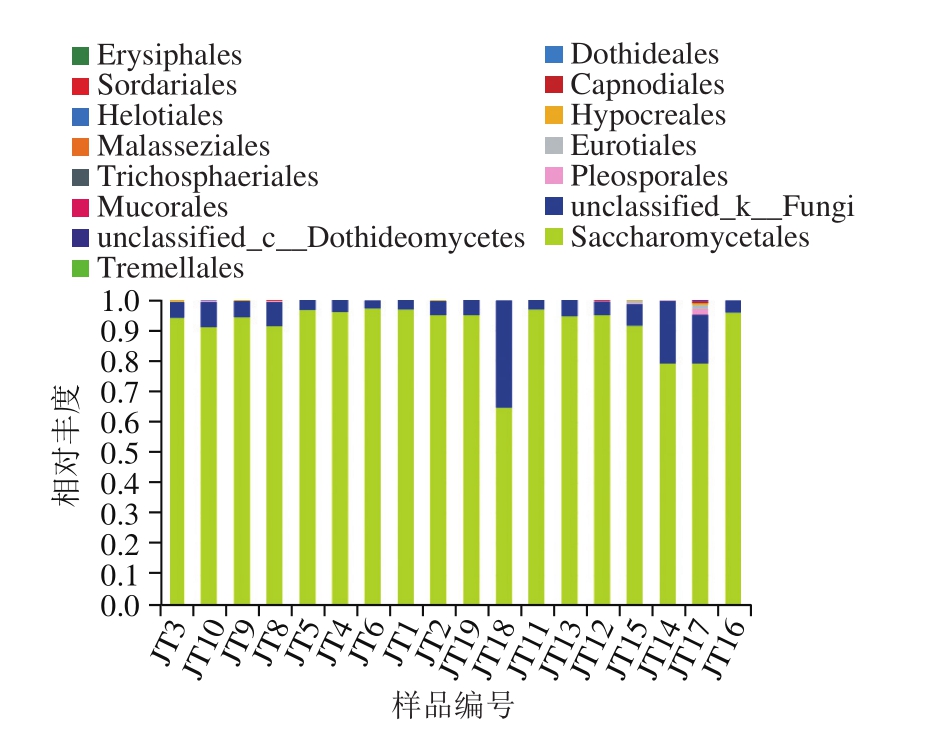
图3 目水平各样品中真菌相对分布柱形图
Fig. 3 Relative abundance of fungi at the order level
在目水平分布见图3,样品中共出现的15 个目,包括13 个已知的目:酵母目(Saccharomycetales)、肉座菌目(Hypocreales)、格孢腔菌目(Pleosporales)、散囊菌目(Eurotiales)、煤炱目(Capnodiales)、座囊菌目(Dothideales)、白粉菌目(Erysiphales)、毛霉目(Mucorales)、马拉色菌目(Malasseziales)、粪壳菌目(Sordariales)、银耳目(Tremellales)、柔膜菌目(Helotiales)、假毛球壳目(Trichosphaeriales),2个未鉴定出:未鉴定出的真菌(unclassified_k__Fungi)和未鉴定出的座囊菌(unclassified_c__Dothideomycetes)。酵母目和unclassified_k__Fungi分别是所有样品中占优势和丰度第2高的目;样品JT3、JT9和JT12中丰度第3是肉座菌目,样品JT6、JT14、JT16、JT17和JT18中丰度第3的是格孢腔菌目,样品JT2和JT15中丰度第3是散囊菌目,样品JT8的丰度第3是煤炱目,样品JT10丰度第3是座囊菌目;另外,白粉菌目、毛霉目、马拉色菌目、粪壳菌目、银耳目、柔膜菌目和假毛球壳目在部分样品中也有很低比例。

图4 科水平各样品中真菌相对分布柱形图
Fig. 4 Relative abundance of fungi at the family level
在科水平分布如图4所示,样品中共出现的25 个科包括17 个已知的科:酵母菌科(Saccharomycetaceae)、发菌科(T r i c h o c o m a c e a e)、假球壳科(Pleosporaceae)、小戴卫霉科(Davidiellaceae)、隐囊菌科(Dothioraceae)、丛赤壳科(Nectriaceae)、复膜孢酵母科(Saccharomycopsidaceae)、肉座菌科(Hypocreaceae)、Trichomonascaceae、白粉菌科(E r y s i p h a c e a e)、毕赤酵母菌科(Pichiaceae)、根霉科(Rhizopodaceae)、马拉色菌科(Malasseziaceae)、毛壳菌科(Chaetomiaceae)、德巴列酵母科(Debaryomycetaceae)、球腔菌科(Mycosphaerellaceae)和毛霉科(Mucoraceae),5 个分类不明:分类不明的肉座菌(norank_o__Hypocreales)、分类不明的银耳菌(norank_o__Tremellales)、分类不明的酵母(norank_o__Saccharomycetales)、分类不明的柔膜菌(norank_o__Helotiales)和分类不明的假毛球壳(norank_o__Trichosphaeriales)和3 个未鉴定出:未鉴定出的真菌(unclassified_k__Fungi)、未鉴定出的酵母菌(unclassified_o__Saccharomycetales)和未鉴定出的座囊(unclassified_c__Dothideomycetes)。酵母菌科和unclassified_k__Fungi占优势和丰度第2;样品JT3、JT9、JT10、JT14和JT18中丰度第3的是norank_o__Saccharomycetales,样品JT2、JT15和JT16中丰度第3的是发菌科,样品JT3丰度第3和第4的分别是norank_o__Hypocreales和发菌科,样品JT6和JT17丰度第3的是假球壳科,样品JT8中丰度第3和第4的分别是小戴卫霉科和假球壳科,样品JT9中丰度第4的是norank_o__Hypocreales,样品JT10中丰度第4和第5的分别是假球壳科和隐囊菌科,样品JT12中丰度第3的是丛赤壳科,样品JT14中丰度第4的是假球壳科,样品JT15中丰度第4的是复膜孢酵母科,样品JT17中丰度第4、第5的分别是发菌科和丛赤壳科,样品JT18中丰度第4的是假球壳科。另外,肉座菌科、Trichomonascaceae、白粉菌科、毕赤酵母菌科、根霉科、马拉色菌科、毛壳菌科、德巴列酵母科、球腔菌科和毛霉科也被发现占较低比例。
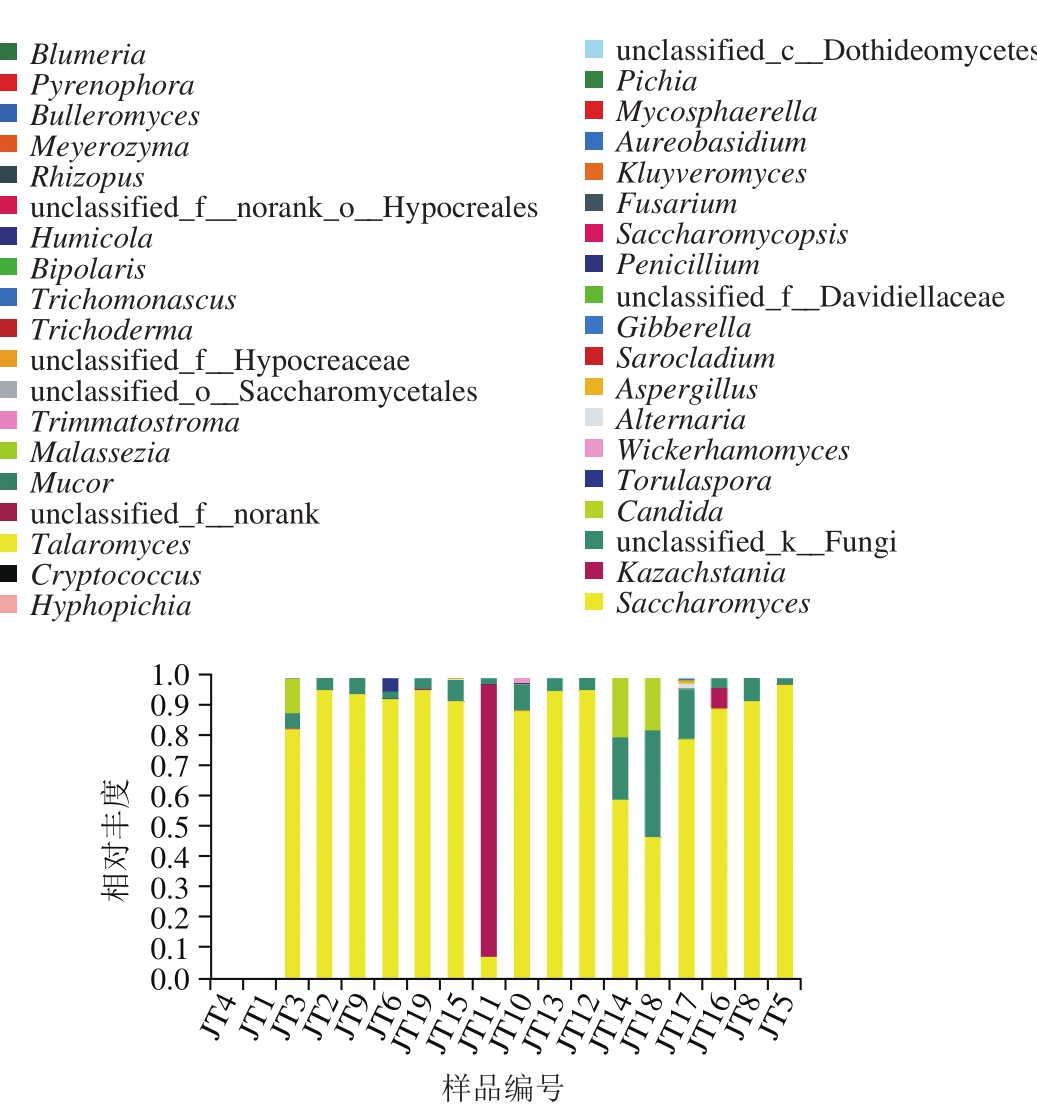
图5 属水平各样品中真菌相对分布柱形图
Fig. 5 Relative abundance of fungi at the genus level
在属水平分布如图5所示,样品中共出现的38 个属包括31 个已知的属:酵母属(Saccharomyces)、哈萨克酵母属(Kazachstania)、假丝酵母属(Candida)、有孢圆酵母属(Torulaspora)、曲霉属(Aspergillus)镰刀菌属(Fusarium)、交链格孢属(Alternaria)、克鲁维氏酵母属(Kluyveromyces)、威克汉姆酵母属(Wickerhamomyces)、帚枝霉属(Sarocladium)、短梗霉属(Aureobasidium)、赤霉属(Gibberella)、复膜孢酵母属(Saccharomycopsis)、毛霉属(Mucor)、木霉属(Trichoderma)、毛红曲霉属(Trichomonascus)、布氏白粉菌属(Blumeria)、平脐蠕孢属(Bipolaris)、丝孢毕赤氏酵母属(H y p h o p i c h i a)、根霉属(Rhizopus)、马拉色菌属(Malassezia)、腐质霉属(Humicola)、布勒担孢酵母属(Bulleromyces)、青霉属(Penicillium)、裸节菌属(Talaromyces)、粉粒座孢属(Trimmatostroma)、隐球酵母属(Cryptococcus)、核腔菌属(Pyrenophora)、短梗霉属(Aureobasidium)、季也蒙毕赤酵母(Meyerozyma)和球腔菌属(Mycosphaerella),6个未鉴定出:未鉴定出的真菌(unclassified_k__Fungi)、未鉴定出的酵母菌(unclassified_o__Saccharomycetales)、未鉴定出的小戴卫霉(unclassified_f__Davidiellaceae)、未鉴定出的座囊菌(unclassified_c__Dothideomycetes)、未鉴定出的肉座菌(unclassified_f__norank_o__Hypocreales)和未鉴定出的肉座菌(unclassified_f__Hypocreaceae),以及其他(Others)。除样品JT11外,酵母属在其余所有样品中都是优势属,而样品JT11优势属是哈萨克酵母属,占88.97%,酵母属只是丰度其第2高的属(7.83%)。即使优势属一致的样品,丰度第2的属也有很大差异,样品JT3丰度第2是假丝酵母属(11.31%),样品JT6丰度第2是有孢圆酵母属(5.31%),样品JT16中丰度第2是哈萨克酵母属(6.72%),其余样品中丰度第2的属都是unclassified_k__Fungi,但在JT11、JT6、JT3、JT16这4个例外样品中丰度第3却是unclassified_k__Fungi;样品JT2丰度第3的是曲霉属(0.20%),样品JT3丰度第4的是曲霉属(0.19%),丰度第5的是镰刀菌属(0.08%),样品JT8丰度第3和第4的属分别是交链格孢属(0.44%)和克鲁维氏酵母属(0.09%),样品JT9中丰度第3和第4的属分别是威克汉姆酵母属(0.68%)和帚枝霉属(0.15%),样品JT10中丰度第3、第4和第5的属分别是威克汉姆酵母属(2%)、假丝酵母属(0.28%)和短梗霉属(0.12%),样品JT12中丰度第3和第4的属分别是赤霉属(0.11%)和假丝酵母属(0.02%),样品JT14中丰度第3和第4的属分别是假丝酵母属(20.01%)和交链格孢属(0.30%),样品JT15中丰度第3和第4的属分别是曲霉属(1%)和复膜孢酵母属(0.16%),样品JT16丰度第4的属是曲霉属(0.11%),样品JT17中丰度第3、第4、第5和第6的属分别是交链格孢属(1.92%)、曲霉属(1.02%)、unclassified_f__Davidiellaceae(0.36%)和毛霉属(0.12%),样品JT18中丰度第3和第4的属分别是假丝酵母属(18.04%)和交链格孢属(0.08%);另外,木霉属、毛红曲霉属、布氏白粉菌属、平脐蠕孢属、丝孢毕赤氏酵母属、根霉属、马拉色菌属、腐质霉属、布勒担孢酵母属、青霉属、裸节菌属、粉粒座孢属、隐球酵母属、核腔菌属、短梗霉属、季也蒙毕赤酵母、球腔菌属在部分样品中也少量出现。

图6 种水平各样本菌群分布热图
Fig. 6 Heatmap of fungi at the species level
在种水平分布见图6,样品中共出现的5 6 个种,包括4 1 个已知的种:酿酒酵母(Saccharomyces cerevisiae)、Kazachstania bulder、光滑假丝酵母菌(Candida glabrata)、戴尔凯氏有孢圆酵母(Torulaspora delbrueckii)、清酒假丝酵母(Candida sake)、异常威克汉姆酵母(Wickerhamomyces anomalus)、萨氏曲霉(Aspergillus sydowii)、玉蜀黍帚枝霉(Sarocladium zeae)、某种交链格孢菌(Alternaria sp.)、近平滑假丝酵母(Candida parapsilosis)、错综赤霉(Gibberella intricans)、出芽短梗霉(Aureobasidium pullulans)、帚状曲霉(Aspergillus penicillioides)、伯顿丝孢毕赤氏酵母(Hyphopichia burtonii)、扣囊复膜孢酵母(Saccharomycopsis fi buligera)、玉蜀黍赤霉(Gibberella zeae)、侵染交链格孢(Alternaria infectoria)、橘青霉(Penicillium citrinum)、近平滑假丝酵母(Candida parapsilosis)、大隐球酵母(Cryptococcus magnus)、矮小假丝酵母(Candida humili)、康宁木霉(Trichoderma koningiopsis)、交织帚枝孢霉(Sarocladium implicatum)、光滑假丝酵母菌(Candida glabrata)、阿曲霉(Aspergillus amstelodami)、Trichomonascus ciferrii、禾布氏白粉菌(Blumeria graminis)、少根根霉(Rhizopus arrhizus)、赤曲霉(Aspergillus ruber)、限制性马拉色菌(Malassezia restricta)、Bulleromyces albus、马克思克鲁维氏酵母(Kluyveromyces marxianus)、黄曲霉(Aspergillus flavus)、黑曲霉(Aspergillus niger)、Trimmatostroma cordae、小麦褐斑长蠕孢霉(Pyrenophora tritici-repentis)、季也蒙毕赤酵母(Meyerozyma guilliermondii)、塔森球腔菌(Mycosphaerella tassiana)、冻土毛霉(Mucor hiemalis)、发酵毕赤酵母(Pichia fermentans)、紧密帚枝霉(Sarocladium strictum)、Aspergillus halophilicu,14 个未鉴定出:未鉴定出的真菌(unclassified_k__Fungi)、某个种真菌(Fungi sp.)、未鉴定出的平脐蠕孢(unclassified_g__Bipolari)、未鉴定出的酵母(unclassified_o__Saccharomycetales)、未鉴定出的镰刀菌(unclassified_g__Fusarium)、未鉴定出的赤霉(unclassified_g__Gibberella)、未鉴定出的座囊菌(unclassified_c__Dothideomycetes)、未鉴定出的小戴卫霉(unclassified_f__Davidiellaceae)、未鉴定的腐质霉(unclassified_g__Humicola)、未鉴定出的肉座菌(unclassified_f__norank_o__Hypocreales)、未鉴定出的肉座菌(unclassified_f__Hypocreaceae)、未鉴定出的木霉(unclassified_g__Trichoderma)、未鉴定出的裸节菌(unclassified_g__Talaromyces)、分类地位未定(Incertae_sedis_sp_g__unclassified)、未鉴定出的青霉(unclassified_g__Penicillium)、未鉴定出的假丝酵母(unclassified_g__Candida)和其他(others)。除样品JT11外,酿酒酵母是其余所有样品中优势种,在样品JT1中最高,达96.7%,在样品JT14和JT18中较低。而在样品JT11的优势种是K. bulder,酿酒酵母属只是其丰度第2高的种。即使优势种都是酿酒酵母的样品,在丰度第2的种上有很大差异,样品JT3中丰度第2的种是光滑假丝酵母菌,样品JT6中丰度第2的种是戴尔凯氏有孢圆酵母,样品JT14中丰度第2是清酒假丝酵母,样品JT16中丰度第2的是K. bulder,其余样品中丰度第2种都是Fungi sp.;但Fungi sp.是JT3、JT6、JT11、JT14、JT16这5 个例外样品中丰度第3的种,样品JT18中丰度第3的是矮小假丝酵母,其余样品丰度第3的种是unclassified_k__Fungi;在随后的丰度第4、第5和第6的种分布上,unclassified_k__Fungi、异常威克汉姆酵母、Alternaria sp.和帚状曲霉出现的频率较高。酿酒酵母、Fungi sp.、unclassified_k__Fungi这3个种在所有样品中都存在,而且数量也较大;K. bulder、戴尔凯氏有孢圆酵母、矮小假丝酵母、光滑假丝酵母菌、清酒假丝酵母这5 种数量较大,但只在单个样品中出现;异常威克汉姆酵母、Alternaria sp.和帚状曲霉数量相对多一些,出现频率较高,在绝大多数样品中都有分布。
2.4 样本比较和物种差异分析

图7 基于非加权UniFrac距离算法的NMDS图
Fig. 7 NMDS based on unweighted UniFrac distance
非度量多维尺度分析(non-metric multi-dimensional scaling,NMDS)是一种将多维空间的研究对象(样本或变量)简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法。其特点是根据样本中包含的物种信息,以点的形式反映在多维空间上,而对不同样本间的差异程度,则是通过点与点间的距离体现的,最终获得样本的空间定位点图。7 个地区、2 种类型共18 个样品基于非加权UniFrac距离算法层级聚类图如图7所示,各样品之间距离较远,但它们之间距离关系趋势和非加权样本层级聚类图很类似。可以看出,与加权UniFrac距离算法相比,非加权UniFrac距离算法聚类较松散,因此,样品之间距离关系被稍作放大,但趋势依然未变,依然也和样本菌群分布bar图和热图结果高度一致。再次印证了样本层级聚类图显示样品中真菌群落结构较相似,与类型和地理分布无关。
3 结论与讨论
本实验采用高通量测序技术从我国不同地区的收集18 个民间面引子样品中发现101 个OTU,属于真菌界4 个门、9 个纲、15 个目、25 个科、38 个属、56 个种;酿酒酵母是大多数样品中的优势菌;酿酒酵母、Fungi sp.、unclassified_k__Fungi这3 个种在所有样品中都存在,而且数量也较大;K. bulder、戴尔凯氏有孢圆酵母、矮小假丝酵母、光滑假丝酵母菌、清酒假丝酵母这5个种数量较大,但只在单个样品中出现;异常威克汉姆酵母、Alternaria sp.和帚状曲霉数量相对多一些,出现频率较高,在绝大多数样品中都有分布。各样品真菌群落结构相差不大,而且在类型和地理分布差异也不明显。
免培养法以环境中的宏基因组或RNA为靶标,采用PCR结合测序技术揭示环境中的微生物多样性,避免了传统分离培养法依赖培养基、培养条件的限制,其结果能更真实地反映样品中国微生物群落结构。PCR-DGGE是早期研究环境中微生物多样性常用的免培养方法之一,张国华[28]基于真菌的ITS区通用引物28S1与5.8S引物采用PCR-DGGE法研究我国小麦主产区甘肃省兰州市、安徽省合肥市、黑龙江省哈尔滨市及山西省临汾市、河南省南阳市的面食发酵剂中真菌菌群结构,共发现杰丁毕赤酵母(Pichia jadinii)、异常毕赤酵母、酿酒酵母、热带假丝酵母(Candida tropicalis)、德尔布孢圆酵母、矮小假丝酵母6 种酵母菌和一种霉菌(米根霉菌(Rhzopus oryzae)),酿酒酵母是优势菌种;吴斯日古冷[29]基于真菌26S rDNA D1区特异引物采用PCR-DGGE法研究内蒙古西部地区7 盟市28 份酸面团中真菌菌群结构,只检测到6 种酵母菌,可能和扩增区域有关。高通量测序是目前最常用的研究环境中微生物多样性的方法,本实验通过高通量测序发现18 份面引子中有56 种真菌,发现的酵母种类比PCR-DGGE更丰富,这也是两种方法的原理所致,但两种方法结果都显示酿酒酵母是大多数样品中的优势菌,说明PCR-DGGE法可以主要用于优势菌和丰度较高的菌研究。
由于Illumina MiSeq测序长度只能在300 bp以内,本实验选取特异性扩增真菌ITS1区测序,长度较普通真菌鉴定用的ITS全长要短的多,再加上分类参考数据库是UNITE数据库,其参考序列较少,因此就会出现本实验结果某个种真菌(Fungi sp.)、未鉴定出的真菌(unclassified_k__Fungi)在所有样品中都存在,而且数量也较大的现象,这些未鉴定到具体种的OTU,有可能是新种,也有可能由于上述技术原因,未比对出结果。
本实验结果发现矮小假丝酵母、光滑假丝酵母菌、清酒假丝酵母在单个样品中出现数量较大,这些菌有些是条件致病菌;Alternaria sp.和帚状曲霉在绝大多数样品中都有分布,数量相对多一些,出现频率较高,这两种真菌都有可能产生毒素;这些具有潜在危险性的菌在面引子中的安全性需要进一步研究。另外,本实验还发现曲霉的其他种、木霉、赤霉、青霉等常见的腐生真菌也少量存在,有可能是污染造成。
本实验用的样品高通量测序结果显示酵母菌群落结构相差不大,而且在类型和地理分布差异不明显,表明面引子中酵母菌群落趋同明显,下一步可以选择更多地区的更多样品分析,进一步印证此结论。
参考文献:
[1] 陈绍军, 吴兆苏. 从我国小麦, 面食及其加工工具的发展历史试谈馒头的起源问题[J]. 农业考古, 1994(1): 219-225.
[2] 滕超, 曲玲玉, 孙伟哲, 等. 传统馒头发酵剂的研究进展[J]. 食品研究与开发, 2015, 36(11): 1-5. DOI:10.3969/j.issn.1005-6521.2015.11.001.
[3] 杨敬雨, 刘长虹. 中国传统酵子的工业化[J]. 食品研究与开发, 2007,28(2): 164-166. DOI:10.3969/j.issn.1005-6521.2007.02.049.
[4] WU C, LIU R, HUANG W, et al. Effect of sourdough fermentation on the quality of Chinese Northern-style steamed breads[J]. Journal of Cereal Science, 2012, 56(2): 127-133. DOI:10.1016/j.jcs.2012.03.007.
[5] 丁长河, 戚光册, 张建华, 等. 传统起子(酵头)的微生物分析及其对馒头品质的影响(英文)[J]. 食品科学, 2007, 28(4): 69-74.DOI:10.3321/j.issn: 1002-6630.2007.04.012.
[6] 韩春然, 马永强, 王金凤, 等. 传统发酵面团的菌相分析[J]. 食品工业科技, 2010, 31(5): 184-187.
[7] HE G Q, ZHANG G H, VUILLAME L, et al. Selection of the optimal association between lactic acid bacteria and yeasts in Chinese sourdoughs[J]. Journal of Pure & Applied Microbiology, 2013, 7(4):3121-3124.
[8] 王雪婷, 廖钰婷, 何瑞, 等. 传统酸面团中优良菌种的筛选、鉴定及在苦荞麸皮馒头中的应用[J]. 食品科技, 2017, 42(2): 156-164.
[9] LIU T J, LI Y, WU S R, et al. Isolation and identification of bacteria and yeast from Chinese traditional sourdough[J]. Modern Food Science & Technology, 2014, 30(9): 114-120; 148.
[10] 刘同杰, 李云, 吴诗榕, 等. 传统酸面团中细菌与酵母菌的分离与鉴定[J]. 现代食品科技, 2014, 30(9): 114-120.
[11] 陈倩. 民间酵面及相关基物中酿酒酵母菌的遗传多样性和群体分化研究[D]. 哈尔滨: 黑龙江大学, 2012.
[12] 潘向辉. 酸面团中酵母菌筛选及发酵特性研究[D]. 保定: 河北农业大学, 2011.
[13] 王冠群, 韩培杰, 杨文菊, 等. 新疆传统发酵乳制品及酵头中酵母菌的分离鉴定[J]. 食品与生物技术学报, 2015, 34(7): 691-698.DOI:10.3969/j.issn.1673-1689.2015.07.004.
[14] 米尔班古丽·阿卜杜如苏力, 玛依古丽·库尔班, 努斯热提古丽·安外尔, 等. 新疆传统馕发酵面团中酵母菌的多样性分析[J].食品科学, 2015, 36(19): 199-203. DOI:10.7506/spkx1002-6630-201519036.
[15] 韩培杰. 中国民间面引子中酿酒酵母遗传多样性和抗逆性研究[D].福州: 福建农林大学, 2013.
[16] 马小红. 馒头老酵子中酵母菌的分离鉴定及其发酵特性分析[D].西安: 陕西师范大学, 2016.
[17] 邓璀. 传统发酵剂中酵母多样性及其在馒头制作中的应用研究[D].郑州: 河南工业大学, 2016.
[18] 熊顺强. 食品基质对酸面团发酵过程中酵母菌和乳酸菌多样性的影响[D]. 南昌: 南昌大学, 2012.
[19] ZHANG J, LIU W, SUN Z, et al. Diversity of lactic acid bacteria and yeasts in traditional sourdoughs collected from western region in Inner Mongolia of China[J]. Food Control, 2011, 22(5): 767-774.DOI:10.1016/j.foodcont.2010.11.012.
[20] ZHANG G, SADIQ F A, ZHU L, et al. Investigation of microbial communities of Chinese sourdoughs using culture-dependent and DGGE approaches[J]. Journal of Food Science, 2015, 80(11):M2535-M2542. DOI:10.1111/1750-3841.13093.
[21] ERCOLINI D. High-throughput sequencing and metagenomics:moving forward in the culture-independent analysis of food microbial ecology[J]. Applied & Environmental Microbiology, 2013, 79(10):3148-3155. DOI:10.1128/AEM.00256-13.
[22] 米其利, 李雪梅, 管莹, 等. 高通量测序在食品微生物生态学研究中的应用[J]. 食品科学, 2016, 37(23): 302-308. DOI:10.7506/spkx1002-6630-201623049.
[23] LITTLEFAIR J E, CLARE E L. Barcoding the food chain: from Sanger to high-throughput sequencing[J]. Genome, 2016, 59(11): 946.DOI:10.1139/gen-2016-0028.
[24] 吴林寰, 陆震鸣, 龚劲松, 等. 高通量测序技术在食品微生物研究中的应用[J]. 生物工程学报, 2016, 32(9): 1164-1174.
[25] 陈星星. 传统发酵酸面团菌群结构的对比及优选菌株益生性能研究[D]. 南昌: 南昌大学, 2015.
[26] BOKULICH N A, MILLS D A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultrahigh-throughput profiling of fungal communities[J]. Applied and Environmental Microbiology, 2013, 79(8): 2519-2526. DOI:10.1128/AEM.03870-12.
[27] KÕLJALG U, NILSSON R H, ABARENKOV K, et al. Towards a unified paradigm for sequence-based identification of fungi[J].Molecular Ecology, 2013, 22(21): 5271.
[28] 张国华. 不同地区传统面食发酵剂中菌群结构及优势菌种代谢的研究[D]. 杭州: 浙江大学, 2014.
[29] 吴斯日古冷. 内蒙古西部地区酸面团中酵母菌和乳酸菌的分离鉴定及其生物多样性研究[D]. 呼和浩特: 内蒙古农业大学, 2011.
Analysis of Fungal Diversity in Homemade Sourdough Starters Using High-Throughput Sequencing
LIU Jianli, SUN Min, CAO Xiaohong, ZHANG Xiu, LI Jingyu
(Key Laboratory of Fermentation and Brewing Engineering and Biotechnology, State Ethnic Affairs Commission,Key Laboratory of Microbial Resources Development and Utilization in Special Habitats, College of Biological Sciences and Engineering,North Minzu University, Yinchuan 750021, China)
Abstract:Sourdough starter is a fermentation starter used in the processing of traditional fl our food. Most of the previous studies concerning the microbiology of sourdough starter have focused on the isolation and culture of microorganisms. This paper aimed to reveal the characteristics of fungal community structure in traditional sourdough starters by high-throughput sequencing. To analyze the fungal communities in 18 samples collected from different provinces in China, internal transcribed spacer 1 (ITS1) regions were amplified with by PCR based on metagenomics and high-throughput sequenced with Illumina MiSeq. Bioinformatics analysis was carried out on Cloud Service Platform. The results showed that a total of 101 operational taxonomic units (OTUs) were found in the 18 samples, which were assigned to 56 species in 38 genera in 25 families in 15 orders in 9 classes in 4 phyla. Saccharomyces cerevisiae was predominant in most samples. S. cerevisiae,Fungi sp. and unclassified_k__Fungi were found abundant in all samples. Kazachstania bulder, Torulaspora delbrueckii,Candida humili, Candida glabrata and Candida sake were also abundant, but they only existed in single samples.Wickerhamomyces anomalus, Alternaria sp. and Aspergillus penicillioides were not only abundant but also highly frequent in most samples. The fungal community structures in these samples were not significantly dissimilar, and no significant difference was observed between the different types and geographical origins.
Keywords:high-throughput sequencing; sourdough starters; fungal community
LIU Jianli, SUN Min, CAO Xiaohong, et al. Analysis of fungal diversity in homemade sourdough starters using highthroughput sequencing[J]. Food Science, 2018, 39(22): 186-194. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201822029. http://www.spkx.net.cn
引文格式:刘建利, 孙敏, 曹晓虹, 等. 利用高通量测序技术分析民间面引子中的真菌多样性[J]. 食品科学, 2018, 39(22): 186-194.DOI:10.7506/spkx1002-6630-201822029. http://www.spkx.net.cn
文章编号:1002-6630(2018)22-0186-09
文献标志码:A
中图分类号:Q938
DOI:10.7506/spkx1002-6630-201822029
第一作者简介:刘建利(1973—),男,副教授,博士,主要从事食品微生物研究。E-mail:LJL7523@126.com
2016年国家级大学生创新训练计划项目(201611407019);发酵酿造工程生物技术国家民委实验室项目(2012-4)
基金项目:北方民族大学重点科研项目(2015KJ36);2017年国家级大学生创新训练计划项目(201711407043);
收稿日期:2017-09-17