郫县年均气温15.7 ℃,相对湿度70%,气候温和,冬无严寒,夏无酷暑,无霜期长。这些环境因素为郫县豆瓣微生物菌群的生存提供了最佳环境,协同优质的蚕豆和二荆条辣椒,凭借300多年的开放式传统酿造技艺,共同造就了郫县豆瓣独特的风味与营养和不可复制的产品特点,也是国家地理标志产品。国内报道了一些关于郫县豆瓣细菌类群的研究,大多为纯培养研究,发现了一些有价值的细菌类群,如乳酸菌、芽孢杆菌等[1-4]。由于纯培养的局限性,一些非培养技术被运用到郫县豆瓣细菌类群的研究中。张琦等[5]运用聚合酶链式反应-变性梯度凝胶电泳技术研究了郫县豆瓣自然发酵过程中细菌群落结构的变化;赵红宇等[6]利用高通量测序技术对郫县豆瓣对发酵过程中细菌群落结构的变化。鉴于开放式的传统发酵工艺中环境的复杂性、污染的不可控性以及产品风味的不稳定性因素,本研究利用高通量测序技术系统全面地研究郫县豆瓣整个发酵期不同阶段的细菌群落及其动态演替规律,以确定不同发酵期细菌优势群体及其可能的污染细菌,为开发郫县豆瓣专用细菌菌剂,保障产品风味质量的稳定性以及安全性提供理论参考和数据支撑。
1 材料与方法
1.1 材料与试剂
实验共采集了郫县豆瓣6 个发酵时期的样本,豆瓣样本编号分别为:BZ1Y、BZ5Y、BZ10Y、peijiao、HE1Y和HE5Y。样品采集使用多点取样法,通过在发酵池的两端以及中间3 个位置分别取上、中、下层等量豆瓣样品,混合均匀备用。6 个时期的样本信息分别为编号BZ1Y、BZ5Y和BZ10Y 3 个样本分别在甜瓣子发酵的第1个月、第5个月和第10个月采样;编号peijiao取样至发酵3 个月的成熟新鲜辣椒醅;编号HE1Y和HE5Y是将发酵10 个月的甜瓣子(BZ10Y)和发酵3 个月的辣椒醅(peijiao)混匀后的发酵阶段,混合发酵时间分别为1 个月和5 个月。
TaqDNA polymerase、dNTPs、DL2000TMDNA Marker 大连宝生物工程有限公司;蛋白酶K 德国Merck公司;溶菌酶 美国Sigma公司;N,N′-亚甲基双丙烯酰胺、去离子甲酰胺 美国Solarbio公司。
1.2 仪器与设备
5430高速冷冻离心机 德国Eppendorf公司;My Cycler型聚合酶链式反应仪 美国Bio-Rad公司;Bio-Best 200E型凝胶成像分析系统 美国西蒙公司;台式冷冻离心机 德国Eppendorf公司;MX-S型可调式混匀仪美国赛洛捷克公司;核酸蛋白微量检测仪 美国MD SpectraMax Drop公司;MiSeq测序仪 美国Illumina公司。
1.3 方法
1.3.1 样品总DNA提取及其高通量测序
采用倪峥飞等[7]的液氮研磨+溶菌酶+十二烷基硫酸钠高盐抽提法提取郫县豆瓣中的细菌总宏基因组,然用核酸蛋白仪检测DNA的浓度和纯度。高通量测序文库的构建和基于Illumina MiSeq平台的测序由金唯智生物科技(北京)有限公司完成。V3-V4可变区采用的测序引物是“CCTACGGRRBGCASCAGKVRVGAAT”和“GGACTACNVGGGTWTCTAATCC”;同时V4-V可变5区采用的测序引物是“GTGYCAGCMGCCGCGGTAA”和“CTTGTGCGGKCCCCCGYCAATTC”。最终得到的序列用于OTU分析,使用VSEARCH进行序列聚类(序列相似性设为97%),用RDP classifier(核酸数据库,Ribosomal Database Program,RDP)贝叶斯算法[8]对OTU的代表性序列进行物种分类学分析。基于OTU的分析结果,采用对样本序列进行随机抽样的方法,分别计算Shannon、Chao1等Alpha多样性指数,并作出稀释曲线。通过Unweighted UniFrac分析比较样本间是否有显著的微生物群落差异,基于Unweighted UniFrac样本间距离矩阵用于主坐标分析(principal co-ordinates analysis,PCoA)可视化2D图展示Beta多样性等。
1.3.2 样品序列的提交及序列注册号获取
通过ENA的FTP服务器,将郫县豆瓣不同发酵时期的样品高通量测序所得fastq序列文件提交到了ENASRA数据库(http://www.ebi.ac.uk/ena/submit),获得其接受号为PRJEB17866。
2 结果与分析
2.1 测序数据统计与OTU分析
按照条形码标签完全匹配方式提取测序序列,高通量测序中通常会出现一些点突变等测序错误,而且序列末端的质量比较低,为了得到更高质量及更准确的生物信息分析结果,需要对测序原始数据进行优化处理。优化处理软件选用Trimmomatic(v0.30 http://www.usadellab.org/cms/?page=trimmomatic)。将上面过滤后的序列与数据库进行比对,去除其中的嵌合体序列,得到最终的有效数据,如表1所示。通过郫县豆瓣不同发酵时期各样品中细菌的16S rRNA基因测序,6 个样品总计测得原始序列条数为1 543 610 条,过滤掉低质量的序列后,总数为1 507 972 条。在上述序列进行去冗余处理后,有效序列数731 188 条,并在97%相似度下将其聚类为用于物种分类的OTU,共产生37 082 个,统计得到所有样品在不同OTU中的丰度信息,各样品测序信息结果如表1所示。peijiao样品中的OTU最多,达到了23 362 个,HE5Y样品中的OTU最少,仅为1 847 个。
表1 不同发酵时期郫县豆瓣样本的测序数据统计
Table1 Statistics of bacterial genomic sequence data for Pixian bean paste samples from different fermentation stages
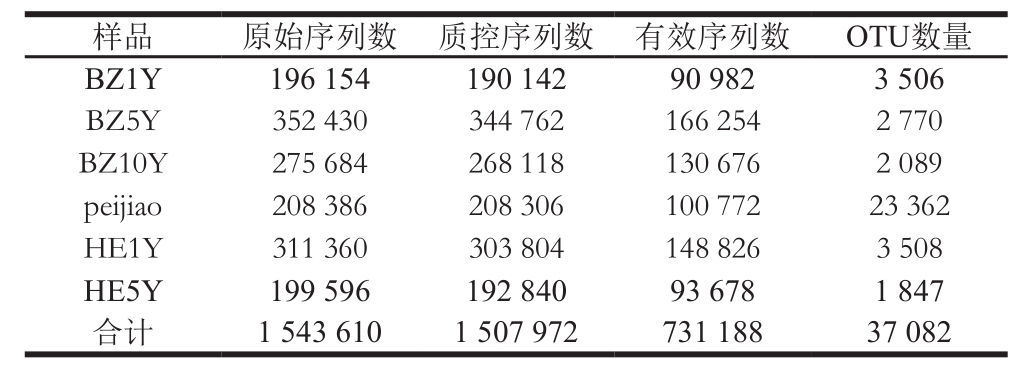
稀释曲线反映了测序的深度,也可以用来评价测序量是否足以覆盖样品所有种群。从图1可知,5个样品(BZ1Y、BZ5Y、BZ10Y、HE1Y和HE5Y)稀释曲线均基本趋于平缓,说明所得序列可基本反映真实环境中细菌群落结构;但样品peijiao呈现一种上升的趋势,很大程度上未达到饱和,说明表明继续测序还可能产生新的OTU。

图1 不同发酵时期郫县豆瓣细菌稀有度曲线
Fig. 1 Rarefaction curves for bacterial communities in Pixian bean paste samples from different fermentation stages
2.2 样品间Alpha多样性分析
表2 样品间Alpha多样性统计
Table2 Alpha diversity of bacterial communities at different fermentation stages
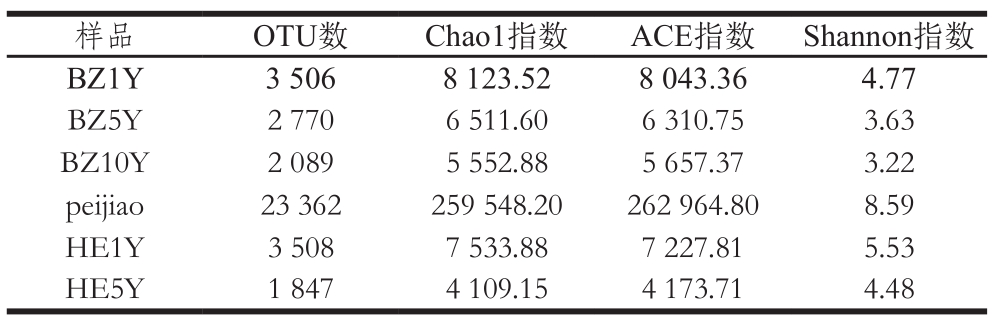
采用Alpha多样性指标中的ACE指数、Shannon指数和Chao1指数对样品序列文库的OUT数目、群落的异质性以及估计群落中物种总数。由表2可以看出,郫县豆瓣不同发酵时期细菌Shannon指数从最初的4.77到发酵结束的4.48,其变化不大,表明样品中群落种群差异性相对较小,但在peijiao发酵期,该指数显著增高,说明该时期的细菌种群差异大。而OTU数、Chao1和ACE指数均明显低于发酵初期(BZ1Y)样品,则表明随着发酵的深入和环境的不断变化,样品中细菌群落的种群多样性和总体丰度不断降低。
2.3 样品间Beta多样性分析
Beta多样性指沿环境梯度不同生境群落之间物种组成的相异性或物种沿环境梯度的更替速率也被称为生境间的多样性。不同群落或某环境梯度上不同点之间的共有种越少,Beta多样性就越大。群落生态学中,Beta多样性主要描述物种组成在时空尺度上的变化。Beta多样性分析可通过多变量统计学方法PCoA实现,直观显示不同环境样品中微生物进化上的相似性及差异性。

图2 样本间细菌群落PCA
Fig. 2 PCA results of microbial communities from Pixian bean paste
主成分分析(principal component analysis,PCA)表明(图2),PC1和PC2分别在样品差异性贡献率上达到72.17%和17.71%,合计达到89.88%,是差异的主要来源。PCA样品BZ1Y、BZ5Y和BZ10Y距离最近,均位于PC2的正值区域,说明三者细菌群落的相似度较高;其次HE1Y与HE5Y相近性次之,样品HE1Y与HE5Y分别处于均位于PC2的负值区域,分别位于PC1坐标轴正侧,较为靠近的位置,说明两样品间的主成分变异不显著;PCA可以看出,5 个样品与peijiao样品的距离较远,细菌群落组相似度较低。总体而言,不同发酵环境及其周期的不同,也显示出样品中细菌群落的分布显著变化。
2.4 样品间门水平的细菌群落结构分析
采用RDP classifier对各样品中的OTU依次进行门、纲、目、科、属的分类信息分析,一共获得11 个门的细菌和一些无法归类的细菌(图3)。这些门分别为Firmicutes、Proteobacteria、Actinobacteria、Chlorobi、Chloroflexi、Gemmatimonadetes、Fusobacteria、Cyanobacteria、Bacteroidetes、Acidobacteria和Deferribacteres。在甜瓣子发酵时期(BZ1Y、BZ5Y和BZ10Y)与辣椒瓣子混合发酵阶段(HE1Y与HE5Y)Firmicutes是优势种群,占到各时期细菌种群的50.7%~90.9%;而在辣椒醅发酵阶段(peijiao)Firmicutes仅占该时期细菌种群的4.2%,而Proteobacteria和Cyanobacteria中的细菌成为peijiao时期发酵的优势群体,分别占全部细菌总数的70.4%和24%。另外,BZ1Y序列分析表明数量较多的细菌群体分别是Firmicutes(80.5%)、Proteobacteria(8.1%)和Actinobacteria(10%);BZ5Y的优势细菌种群是Firmicutes(86.5%)、Proteobacteria(3%)和Actinobacteria(9.7%);发酵10 个月后,优势群体的种类没有改变,但数量已经发生了变化,BZ10Y中的优势细菌Firmicutes、Proteobacteria和Actinobacteria的含量分别变化为90.9%、5.4%和2.7%。由于辣椒醅混入豆瓣发酵,带入了新的细菌类群,因此在混合1个月发酵(HE1Y)后优势细菌成为Firmicutes(52.7%)、Cyanobacteria(10.6%)、Proteobacteria(30.7%)和Actinobacteria(5.7%);混合发酵豆瓣成熟后(HE5Y)优势细菌为Firmicutes(70.7%)、Cyanobacteria(17.1%)、Proteobacteria(6.4%)和Actinobacteria(5.5%)。说明郫县豆瓣整个发酵过程中,细菌种群始终处于动态的变化当中。
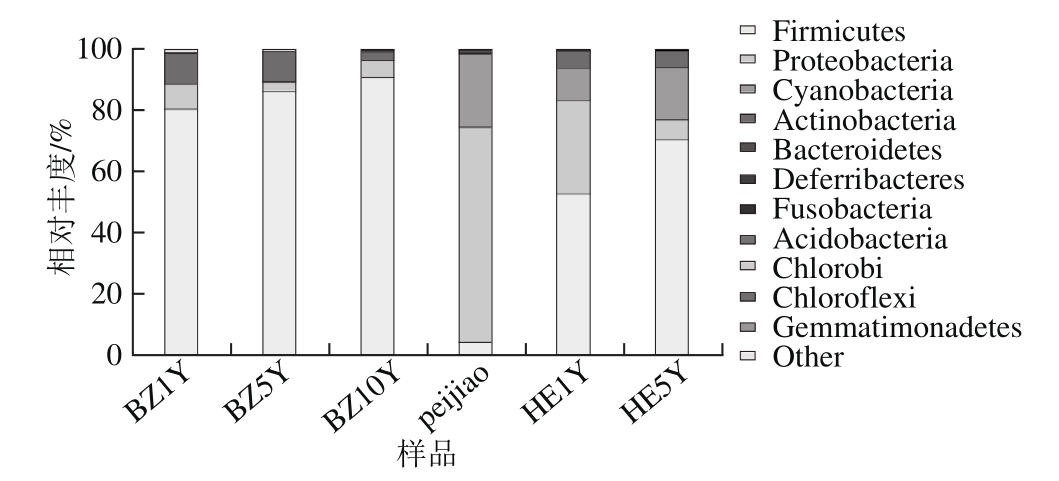
图3 门水平各样本菌群分布图
Fig. 3 Phylum frequencies of microbial communities from samples
2.5 样品间属水平的细菌群落分布
通过高通量测序,郫县豆瓣整个发酵过程中共有185 个属的细菌和部分未能分类的细菌被挖掘。属水平的细菌群落数量在前50的属热图分析(图4)表明,测序结果中组成比例较高的前50 个属分别是Hafnia、Methylobacterium、Paracoccus、Chromohalobacter、Halanaerobium、Halomonas、Ochrobactrum、Rhizobium、Rahnella、Wohlfahrtiimonas、Pectobacterium、Tatumella、Empedobacter、Klebsiella、Myroides、Acinetobacter、Stenotrophomonas、Serratia、Lampropedia、Alcaligenes、Brachybacterium、Leucobacter、Providencia、Brevibacterium、Ignatzschineria、Enterococcus、Arthrobacter、Lactococcus、Jeotgalicoccus、Kurthia、Proteus、Morganella、Paenochrobactrum、Lentibacillus、Oceanobacillus、Gracilibacillus、Virgibacillus、Staphylococcus、Pantoea、Enterobacter、Pseudomonas、Brenneria、Leuconostoc、Vagococcus、Weissella、Tetragenococcus、Lactobacillus、Pediococcus、Corynebacterium和Bacillus。热图分析表明了郫县豆瓣不同发酵时期样品间不同细菌属的相对丰度以及细菌组成的差异性和和样品间的相似性。如热图中的BZ1Y样品中检测到38 个属,BZ5Y中37 个属,BZ10Y中36 个属;单独发酵成熟的peijiao样品中检测到44 个属,由于辣椒醅的混入,瓣子与辣椒醅的混合发酵HE1Y样品中有47 个属,随着混合发酵的继续进行,在成品豆瓣HE5Y样品中获得42 个属。样品间属水平的聚类关系(图4)可以看出BZ1Y、BZ5Y和BZ10Y聚在一起,表明3个样品的相似度较高;HE1Y与HE5Y的相似度也较高,而peijiao样品形成了一个独立的分支,说明peijiao与其他5 个样品的相似度较低,与其他5 个样品的细菌组成差异较大。这个结果也符合郫县豆瓣的实际发酵情况,因为peijiao样品是单独发酵的,与甜瓣子的发酵环境、材料等差异较大。
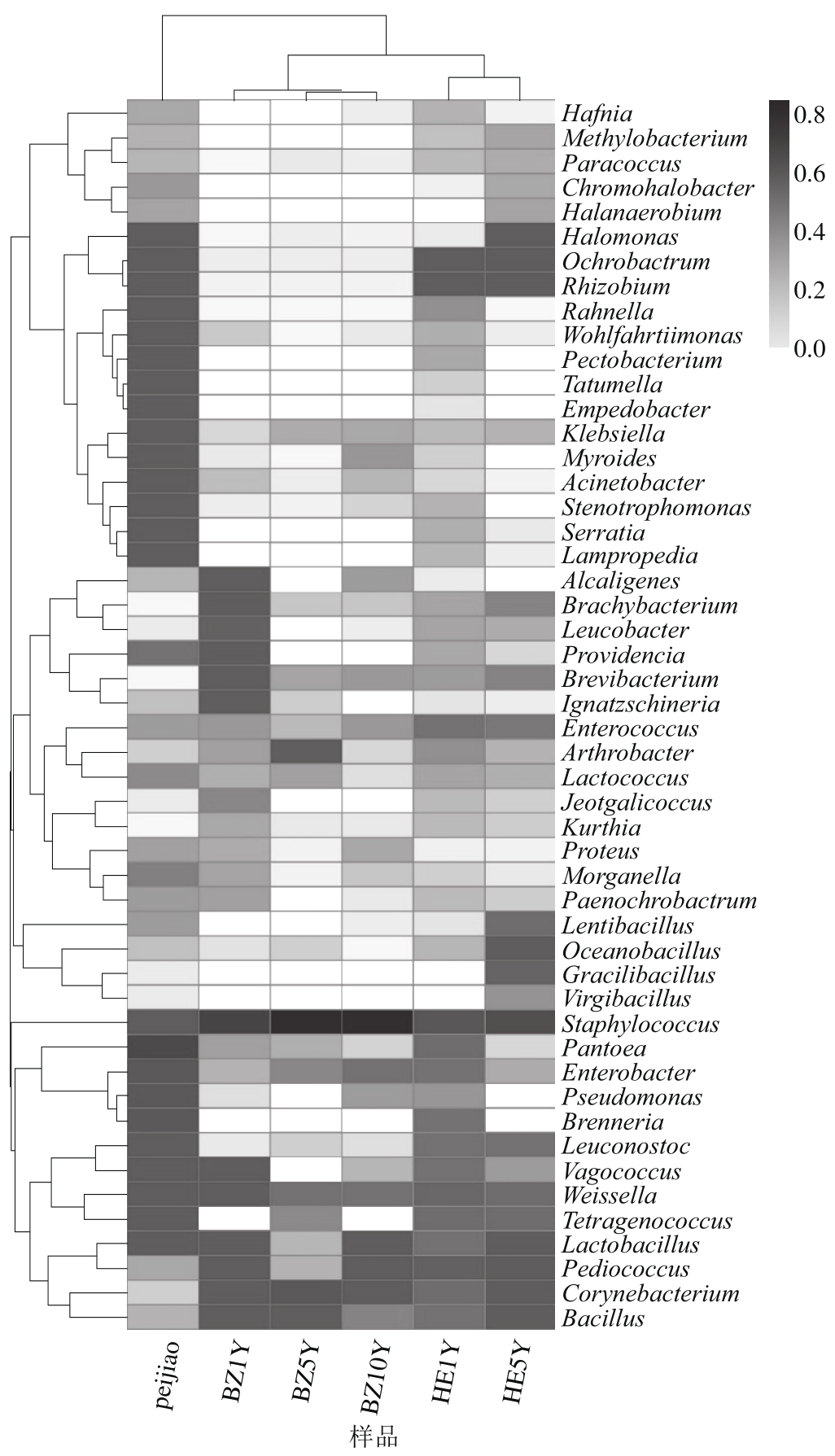
图4 不同样品间的细菌热图
Fig. 4 Heat map of bacteria in different samples
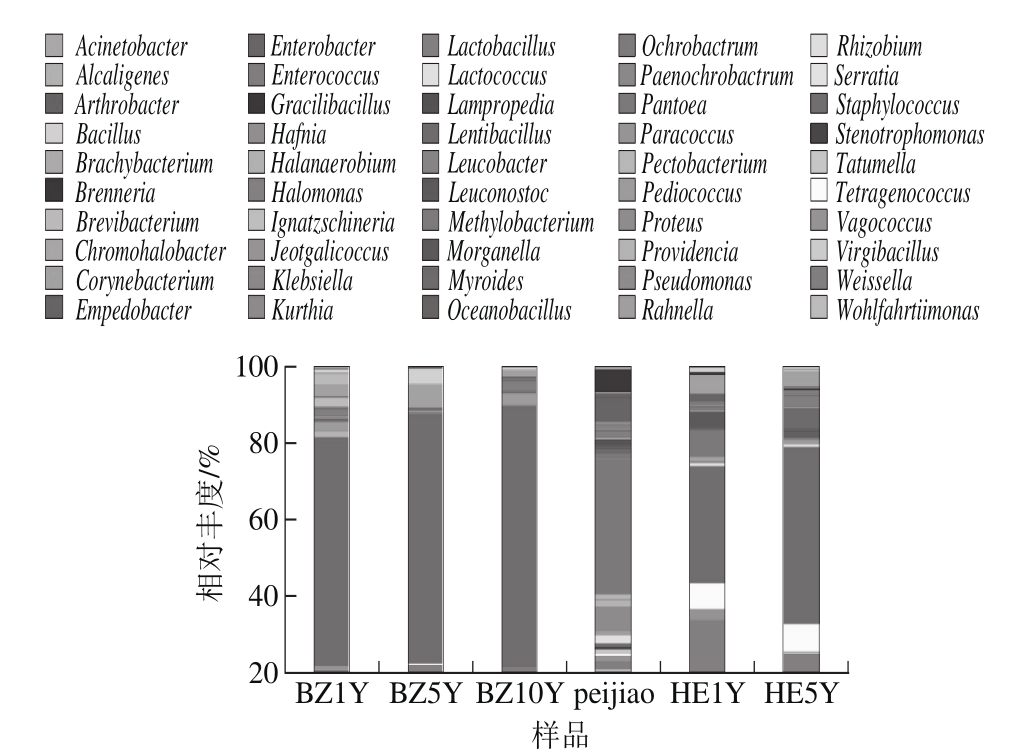
图5 各样品在属水平的细菌群落结构组成差异
Fig. 5 Bacterial community structures of all the samples at the genus level
图5 反映了不同样品间属水平细菌(前50)群落组成的变化情况。在瓣子发酵初期(BZ1Y)的主要优势细菌为Staphylococcus(65.31%)、Corynebacterium(3.45%)、Brevibacterium(3.75%)、Ignatzschineria(2.46%)、Lactobacillus(2.26%)和Pediococcus(2.24%)。随着发酵的进行Staphylococcus的含量逐渐上升,Corynebacterium数量先上升后下降,Brevibacterium的含量呈现下降的趋势。在瓣子发酵的中期(BZ5Y)的主要细菌为Staphylococcus(74.07%)、Corynebacterium(6.99%)、Bacillus(4.24%)和Weissella(2.49%),Bacillus和Weissella的细菌出现大幅增长。同时,也存在着多种新物种的出现和初始物种的消失,如Jeotgalicoccus是瓣子发酵初期的原始物种,而到了发酵后期未被检测到。Brenneria和Methylobacterium出现在瓣子发酵的中期,但在发酵初期和发酵后期均未检测到;而Halomonas和Paracoccus也出现在瓣子发酵中期,随后一直存伴随于瓣子的发酵过程。到了瓣子发酵的末期(BZ10Y),优势菌群为Staphylococcus(79.93%)、Pediococcus(3.28%)、Lactobacillus(3.22%)、Corynebacterium(2%)和Weissella(2%)。此时,同步单独发酵的辣椒醅(peijiao)成品中的优势细菌为Pantoea(13.08%)、Pseudomonas(2.32%)、Enterobacter(2.26%)和Brenneria(2.16%),并且有高达44.72%的物种未归类,这些物种可能存在一些新的物种信息,值得深入研究。在辣椒醅与瓣子混合的初期HE1Y中优势菌为Staphylococcus(25.26%)、Weissella(11.51%)、Tetragenococcus(5.57%)、Pantoea(5.35)和Corynebacterium(3.78%);到了发酵末期,成熟豆瓣HE5Y中的优势细菌变为Staphylococcus(43.81%)、Tetragenococcus(6.81%)、Weissella(4.89%)、Corynebacterium(3.41%)和Lactobacillus(3.09%)。因此,郫县豆瓣的发酵过程中伴随着优势种群的数量和种类的动态变化,同时也影响着产品的质量与风味。
3 讨 论
目前,已经发现的纯培养微生物还不到自然界微生物总量的1%[9],因此,利用传统分离技术全面揭示样品中的微生物组成几乎不可能。宏基因组学于1998年提出[10],随后高通量测序技术的出现不仅使得环境微生物被全貌解析成为可能[11];近年来,高通量测序技术也广泛的应用于水环境、陆地、肠道、动植物等环境微生物检测[12-18],而且在在环境监测、生产管理、微生物疾病控制和生态评估等方面发挥着重要意义[19-21]。
高通量测序技术的快速发展对食品微生物发酵过程和机制研究产生了深刻的影响,对食品发酵工程优化提供了很好的数据支撑[22-26]。郫县豆瓣作为我国著名的传统发酵食品,开放式的半固态发酵体系和适宜的郫县气候条件高效的网罗了大量环境细菌参与豆瓣酿造。这些环境中的细菌,随着发酵时间的推移而发生着显著的动态变化。同时,悠久的酿造历史也使得一些细菌在高盐浓度条件下逐渐适应,并成为郫县豆瓣中的优势种群,为郫县豆瓣风味和质量的稳定性保驾护航。本研究完成了郫县豆瓣全发酵过程的细菌高通量测序,结果表明伴随于整个郫县豆瓣发酵的优势细菌属为:Staphylococcus、Weissella、Pediococcus、Lactobacillus、Corynebacterium和Bacillus,它们在数量上随发酵时间有所变化,但相对比较稳定;毫无疑问,它们对郫县豆瓣的质量与风味产生重要的影响。这些研究成果也为未来开发郫县豆瓣专用细菌菌剂提供了很好的参考价值。张琦等[5]采用聚合酶链式反应-变性梯度凝胶电泳对郫县豆瓣细菌的研究发现Staphylococcus xylosus是整个发酵过程中的优势细菌,本研究中Staphylococcus中的细菌数量(25%~80%)也在整个发酵过程中占据绝对优势。通常Staphylococcus中的金黄色葡萄球菌为致病菌,表皮葡萄球菌中有部分是致病菌,S. xylosus是弱致病性的,而腐生葡萄球菌是不致病的。葡萄球菌属的细菌部分具有耐盐性,生长营养需求不高,可能将导致豆瓣酱成品质量检测不合格。Staphylococcus在郫县豆瓣发酵过程中具体起着有益的还是有害的作用都需进一步深入探索。因此,在生产中需要多加关注Staphylococcus菌群的生产控制,在未来研发中要阐明此类菌群的功能性。Bacillus也是郫县豆瓣的优势种群,这与笔者前期的纯培养结果[1,3,27]以及赵红宇等[6]的实验结果基本一致。另外,首次发现Pediococcus和Corynebacterium作为郫县豆瓣发酵过程的优势菌。据研究[28-30]Pediococcus在酿酒环境中经常被发现该菌在豆瓣中的功能还需要进一步探索,以便于更好地为郫县豆瓣产业的规模化和现代化服务。
参考文献:
[1] 董丹, 关统伟, 赵辉平, 等. 两个不同发酵时期豆瓣中微生物多样性的差异对比[J]. 中国酿造, 2014, 33(11): 55-58. DOI:10.11882/j.issn.0254-5071.2014.11.012.
[2] ZOU Y L, XIANG W L, RAO Y, et al. Diversity analysis of bacteria in the latter ripening of Pixian soybean paste fermentation based on 16S rDNA analysis[J]. African Journal of Microbiology Research, 2012, 6(42):7008-7012. DOI:10.5897/AJMR12.1341.
[3] 赵辉平, 关统伟, 董丹, 等. 郫县豆瓣中可培养细菌多样性分析及酶活性初筛[J]. 中国调味品, 2015, 40(8): 5-9. DOI:10.3969/j.issn.1000-9973.2015.08.002.
[4] 刘超兰, 黄著, 彭熙敏, 等. 乳酸菌和酵母共培养技术缩短郫县豆瓣酱陈酿期的应用研究[J]. 中国酿造, 2009, 28(3): 105-108.DOI:10.3969/j.issn.0254-5071.2009.03.032.
[5] 张琦, 汪先丁, 杨虎, 等. 郫县豆瓣自然发酵过程中细菌群落结构的变化[J]. 食品与发酵科技, 2010, 46(6): 16-18. DOI:10.3969/j.issn.1674-506X.2010.06.005.
[6] 赵红宇, 徐炜桢, 杨国华, 等. 基于高通量测序的郫县豆瓣后发酵期细菌多样性研究[J]. 食品科学, 2017, 38(10): 117-122. DOI:10.7506/spkx1002-6630-201710020.
[7] 倪峥飞, 许伟, 窦文芳, 等. 镇江香醋固态发酵醋醅中微生物总DNA提取方法比较[J]. 微生物学报, 2010(1): 119-125. DOI:10.13343/j.cnki.wsxb.2010.01.011.
[8] WANG Q, GARRITY M G, TIEDIE M J, et al. Bayesianclassifier for rapid assignment of rRNA sequencesinto the new bacterial taxonomy[J].Applied and Environmental Microbiology, 2007, 73(16): 5261-5267.DOI:10.1128/AEM.00062-07.
[9] AMANN R I, LUDWIG W, SCHLEIFER K H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J]. Microbiological Reviews, 1995, 59(1): 143-169.
[10] HANDESMAN J, RONDON M R, BRADY S F, et al. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products[J]. Chemistry & Biology, 1998, 5(10):245-249. DOI:10.1016/S1074-5521(98)90108-9.
[11] GLENN T C. Field guide to next-generation DNA sequencers[J].Molecular Ecology Resources, 2011, 11(5): 759-769. DOI:10.1111/j.1755-0998.2011.03024.x.
[12] 叶雷, 闫亚丽, 陈庆森, 等. 高通量测序技术在肠道微生物宏基因组学研究中的应用[J]. 中国食品学报, 2016, 16(7): 216-223.DOI:10.16429/j.1009-7848.2016.07.029.
[13] TURNBAUGH P J, RIDAURA V K, FAITH J J, et al. The effect of diet on the human gut microbiome: a metagenomicanalysis in humanized gnotobioticmice[J]. Science Translational Medicine, 2009,1(6): 6799-6806. DOI:10.1126/scitranslmed.3000322.
[14] ZHANG C H, ZHANG M H, WANG S Y, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice[J]. The ISME Journal, 2009, 4(2): 232-241. DOI:10.1038/ismej.2009.112.
[15] 葛英亮, 于水利, 时文歆, 等. 应用Illumina MiSeq高通量测序技术解析O3-BAC饮用水处理过程细菌多样性变化[J]. 食品科学, 2016,37(16): 223-228. DOI:10.7506/spkx1002-6630-201616036.
[16] ARFI Y, BUÉE M, MARCHAND C, et al. Multiple markers pyrosequencing reveals highly diverse and host- specific fungal communities on the mangrove trees Avicennia marina and Rhizophora stylosa[J]. FEMS Microbiology Ecology, 2012, 79(2): 433-444.DOI:10.1111/j.1574-6941.2011.01236.x.
[17] 王佩雯, 朱金峰, 陈征, 等. 高通量测序技术下连作植烟土壤细菌群落与土壤环境因子的耦合分析[J]. 农业生物技术学报, 2016, 24(11):1754-1763. DOI:10.3969/j.issn.1674-7968.2016.11.013.
[18] 李旭光, 齐占会, 林琳, 等. 基于高通量测序分析的大鹏澳海域沉积物古菌群落结构初步研究[J]. 南方水产科学, 2015(6): 1-8.DOI:10.3969/j.issn.2095-0780.2015.06.001.
[19] TABERLET P, COISSAC E, POMPANON F, et al. Towards next-generation biodiversity assessment using DNA metabarcoding[J]. Molecular Ecology, 2012,21(8): 2045-2050. DOI:10.1111/j.1365-294X.2012.05470.x.
[20] YU D W, JI Y Q, EMERSON B C, et al. Biodiversity soup:metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring[J]. Methods in Ecology and Evolution, 2012, 3(4): 613-623. DOI:10.1111/j.2041-210X.2012.00198.x.
[21] JI Y Q, ASHTON L, PEDLEY S M, et al. Reliable, verifiable and efficient monitoring of biodiversity viametabarcoding[J]. Ecology Letters,2013, 16(10): 1245-1257. DOI:10.1111/ele.12162.
[22] 吴林寰, 陆震鸣, 龚劲松, 等. 高通量测序技术在食品微生物研究中的应用[J]. 生物工程学报, 2016, 32(9): 1164-1174. DOI:10.13345/j.cjb.150552.
[23] WOLFE B E, BUTTON J E, SANTARELLI M, et al. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity[J]. Cell, 2014, 158(2): 422-433. DOI:10.1016/j.cell.2014.05.041.
[24] LIU W, XI X, SUDU Q, et al. High-throughput sequencing reveals microbial community diversity of Tibetan naturally fermented yak milk[J]. Annals of Microbiology, 2015, 65(3): 1741-1751.DOI:10.1007/s13213-014-1013-x.
[25] 邓杰, 黄治国, 卫春会, 等. 基于高通量测序的浓香型白酒窖池细菌群落结构分析[J]. 现代食品科技, 2015, 31(7): 50-55. DOI:10.13982/j.mfst.1673-9078.2015.8.033.
[26] 左勇, 王小龙, 叶碧霞, 等. 基于高通量测序对宜宾芽菜中细菌群落结构分析[J]. 食品工业科技, 2016, 37(10): 242-250. DOI:10.13386/j.issn1002-0306.2016.10.040.
[27] 董丹, 关统伟, 车振明, 等. 发酵初期豆瓣酱中微生物多样性分析及产酶菌株的筛选[J]. 食品工业, 2015, 36(7): 175-178.
[28] ABRUNHOSA L, INES A, RODRIGUES A I, et al. Biodegradation of ochratoxin A by Pediococcus parvulus isolated from Douro wines[J]. International Journal of Food Microbiology, 2014, 188: 45-52. DOI:10.1016/j.ijfoodmicro.2014.07.019.
[29] JORGE H G G, LUIS C D B, JUAN C C A, et al. Pediococcus damnosus strains isolated from a brewery environment carry the horA gene[J]. Institute of Brewing & Distilling, 2017, 123(1): 77-80.DOI:10.1002/jib.397.
[30] WALLING E, GINDREAU E, LONVAUD-FUNEL A.Exopolysaccharide biosynthesis by Pediococcus damnosus strains isolated from wine: elaboration of molecular detection tools[J]. Lait,2001, 81(1): 289-300. DOI:10.1051/lait:2001132.