葡萄酒的风格不仅取决于酿酒葡萄品种、酿造工艺,而且与发酵过程中微生物的代谢息息相关。新西兰科学家研究表明,决定葡萄酒独特风味的化合物中,大约有一半来自微生物的代谢过程。在自然发酵过程中,葡萄酒中的微生物主要来源酿酒葡萄、葡萄园、酿造设备、酒厂车间环境等[1-4],这些微生物在发酵过程中会随着葡萄果粒和酿造工序进入发酵罐参与发酵过程,因此对葡萄酒的品质形成有一定影响。
酿酒葡萄在生长过程中,受葡萄园土壤类型、地理地形、光照、降水量、昼夜温差等自然因素的影响,附着在葡萄原料表皮中的微生物种类存在显著差异[5]。目前世界上许多葡萄酒产地的本土微生物菌群已经被广泛研究,如西班牙、意大利、法国、美国、澳大利亚等。我国葡萄栽培区域广阔,葡萄适栽区的生态地理条件复杂多样,势必蕴藏着丰富的酿酒微生物菌资源。但遗憾的是除酿酒葡萄自然发酵液中酵母多样性的分析引起研究人员的极大关注外[6-9],对于葡萄表面微生物资源和葡萄园中微生物的生态研究一直未得到应有的重视,其多样性研究在我国尚属空白。
本研究利用高通量测序技术分析新疆产区3 个不同地区葡萄园中酿酒葡萄、土壤以及葡萄叶片中微生物的组成,揭示同种生态系统中地生微生物群落和附生微生物群落之间、不同生态系统微生物群落之间的生态联系及交互作用,为新疆产区酿酒微生物资源库的建设以及优良微生物的筛选提供基础。
1 材料与方法
1.1 材料与试剂
实验样品来自中国新疆天山北麓酿酒葡萄产区中的A、B和C 3 个葡萄园中的酿酒葡萄(G)、葡萄叶片(L)和葡萄园土壤(S),分别于2015年9月采样,每种样品采集3 份,样品具体如表1所示。
表1 实验样品
Table 1 Experimental samples
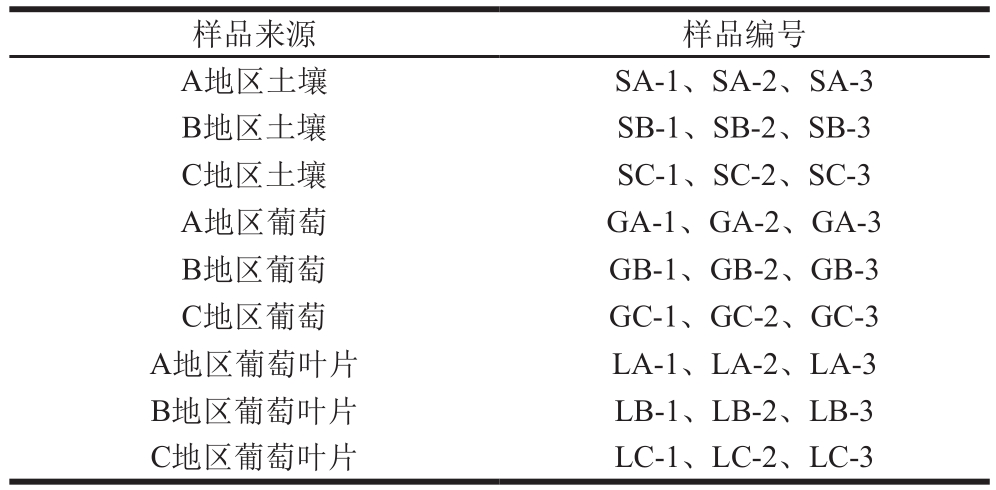
 A地区土壤 SA-1、SA-2、SA-3 B地区土壤 SB-1、SB-2、SB-3 C地区土壤 SC-1、SC-2、SC-3 A地区葡萄 GA-1、GA-2、GA-3 B地区葡萄 GB-1、GB-2、GB-3 C地区葡萄 GC-1、GC-2、GC-3 A地区葡萄叶片 LA-1、LA-2、LA-3 B地区葡萄叶片 LB-1、LB-2、LB-3 C地区葡萄叶片 LC-1、LC-2、LC-3
A地区土壤 SA-1、SA-2、SA-3 B地区土壤 SB-1、SB-2、SB-3 C地区土壤 SC-1、SC-2、SC-3 A地区葡萄 GA-1、GA-2、GA-3 B地区葡萄 GB-1、GB-2、GB-3 C地区葡萄 GC-1、GC-2、GC-3 A地区葡萄叶片 LA-1、LA-2、LA-3 B地区葡萄叶片 LB-1、LB-2、LB-3 C地区葡萄叶片 LC-1、LC-2、LC-3琼脂糖 西班牙Biowest Agarose公司;NGS-ITS1 copy1 Barcode1-70、NGS-16s V4 copy1 Barcode1-70生工生物工程(上海)股份有限公司;Gold View I核酸染料 北京中生瑞泰科技有限公司;DL 2000 Marker美国Axygen公司;所有提取用无机、有机溶剂均为国产分析纯。
1.2 仪器与设备
Illumina Misq测序平台 国家级酒类品质与安全国际联合研究中心;电子天平 瑞士梅特勒-托利多公司;立式电热压力蒸汽灭菌锅 上海申安医疗器械厂;2-16N高速微量离心机 湖南恒诺仪器设备有限公司;涡旋振荡器 美国Scientific Industries公司;Mini-beater组织研磨器 美国Biospec公司;BioSpec-nano核酸蛋白检测仪 德国Eppendorf公司;BG-Power 600电泳仪郑州南北仪器设备有限公司;Biosystems Model温度梯度聚合酶链式反应(polymerase chain reaction,PCR)仪、Tanon 1600凝胶成像仪 美国伯乐公司。
1.3 方法
1.3.1 样品采集
土壤采样方法:去除葡萄地表面的植物等残体,用土铲垂直切开土壤,在土壤深度20 cm处取样,每个取样点取样约0.5 kg[10]。
葡萄叶片、葡萄采样方法:以一株葡萄树的上、中、下3 个部位采样,用剪刀剪下葡萄叶片和葡萄串[11]。
1.3.2 样品细菌和真菌的分离
1.3.2.1 土壤细菌和真菌的分离
将块状土壤轻轻破碎为粉末状,称取0.4 g土壤样品和0.5 g研磨珠于灭菌的2 mL离心管中。
1.3.2.2 葡萄及葡萄叶片细菌和真菌的分离[12-14]
1)称取葡萄皮、叶片(无果肉、无果梗)各4 g,加入到灭菌的50 mL离心管L1中,加6 mL TENP缓冲液(50 mmol/L Tris,20 mmol/L EDTA,100 mmol/L NaCl,1% PVP,pH 10.0),涡旋振荡10 min;2)配平,3 000×g离心5 min,将上清液转移至一个新的50 mL离心管L2中;3)再吸取6 mL TENP缓冲液到离心管L1中,涡旋振荡5 min,配平,3 000×g离心5 min,将上清液转移至离心管L2中;4)重复步骤3);5)吸取6 mL磷酸盐缓冲液(phosphate buffer saline,PBS)到离心管L1中,涡旋振荡5 min,配平,3 000×g离心5 min,将上清液转移至离心管L2中;6)将离心管L2配平,9 000×g离心10 min,弃上清液,留下的沉淀即为所需样品,如需放置则-20 ℃保存。
1.3.3 DNA的粗提
土壤中微生物原基因组DNA的提取采用Mag-Bind Soil DNA Kit Protocol试剂盒(OMEGA);葡萄表面、葡萄叶片微生物原基因组DNA的提取采用Fast DNA SPIN Kit for Soil试剂盒(MP)。
1.3.4 样品分析
由中国食品发酵工业研究院国家级酒类品质与安全国际联合研究中心应用Illumina Misq测序平台进行分析。
1.3.4.1 样品PCR[15-16]
真菌ITS ITS1区域扩增通用引物:ITS 1F(5’-CTTGGTCATTTAGAGGAGTAA-3’);ITS 1R(5’-GCTGCGTTCTTCATCGATGC-3’)。PCR(50 μL)扩增体系:dNTP Mixture 4 μL,10×PCR Buffer(Mg2+plus)5 μL,正向和反向引物各1 μL,模板DNA 5 μL,Ex Taq酶0.25 μL,ddH2O补足至50 μL,充分混匀。PCR扩增程序:98 ℃预变性3 min;98 ℃变性45 s,53 ℃退火30 s,72 ℃延伸45 s,反应35 个循环;72 ℃延伸8 min。-20 ℃保存。
细菌16S rDNA V4扩增通用引物:16S 515F(5’-GTGCCAGCMGCCGCGGTAA-3’);16S 806R(5’-GGACTACHVGGGTWTCTAAT-3’)。PCR(50 μL)扩增体系:dNTP Mixture 4 μL,10×PCR Buffer(Mg2+plus) 5 μL,正向和反向引物各1 μL,模板DNA 5 μL,Ex Taq酶0.25 μL,ddH2O补足至50 μL,充分混匀。PCR扩增程序:95 ℃预变性3 min;95 ℃变性45 s,50 ℃退火30 s,72 ℃延伸1 min,反应35 个循环;72 ℃延伸5 min。-20 ℃保存。
1.3.4.2 PCR产物琼脂糖凝胶电泳分析
采用DL2000 Marker、1%的琼脂糖凝胶,110 V恒压对PCR产物进行电泳35 min,凝胶成像仪观察电泳结果。
1.3.5 高通量测序[17-23]
使用AxyPrep DNA凝胶回收试剂盒对真菌、细菌的PCR扩增产物进行切胶回收。采用Life Qubit 3.0对纯化后的PCR产物进行浓度定量,构建真菌ITS ITS1、细菌16S rDNA V4区域文库。将样品(文库)逐步稀释到4 nm,按1∶1加入氢氧化钠室温变性5 min,加入HT1 Buffer预冷,选8 ppm进行高通量上机测序。
1.3.6 原始测序数据统计
下机后,采用双峰(pair-end)测序法舍弃原始数据中的低质量序列(保证50 个连续碱基的平均质量大于Q30)。用Flash软件,过滤对接上的序列(连续相同的碱基个数小于6,模糊碱基个数小于1),设计无法对接的序列,得到最终用于OTU(operational taxonomic unit)分析的序列。
1.3.7 OTU列表生成
采用Qiime软件,将相似度达97%以上的序列归为一个操作分类单元即OTU;聚类所有序列(Ucl法),参照RDP(Ribosomal Database Project)数据库,采用贝叶斯算法注释每个分类中的OTU代表序列,得到每个OTU的分类学信息。
1.3.8 微生物群落结构及多样性分析
对OTU数据进行整理,在微生物分类为属的水平上对各个样本中的真菌、细菌进行物种丰度柱形图统计,确定优势菌属;通过主坐标分析图及聚类图分析样本间相似度;通过维恩图观察A、B、C 3 个葡萄园中样本间所包含的相同菌属,以此来综合分析各个样本中微生物群落的多样性。
2 结果与分析
2.1 实验样品中微生物群落多样性结果
高通量测序结果表明,本研究27 个样品中共检测出了272 种不同属的真核微生物,属于Ascomycota、Basidiomycota、Chytridiomycota、Zygomycota和Un—s-Fungal sp 38 CC 06_28这5 个菌门;检测出了317 种原核微生物,属于Actinobacteria、Bacteroidetes、Crenarchaeota、Firmicutes、Nitrospirae、Planctomycetes、Proteobacteria和Verrucomicrobia这8 个菌门。含量较多的前15 种菌属如表2、3所示。
表2 3 个不同地区土壤、葡萄和叶片中属水平真菌微生物数量
Table 2 Fungal quantities at family level in soil, as well as on grapes and leaves
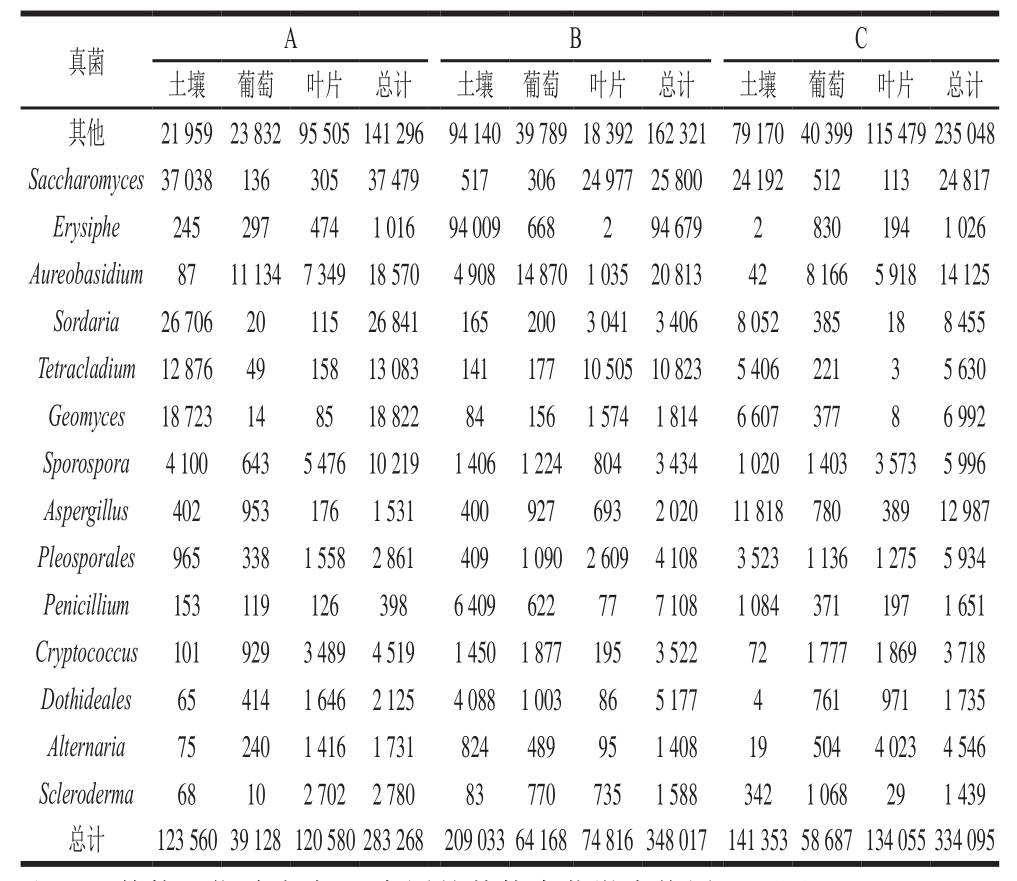
注:“其他”指除表中14 个属外其他真菌微生物属。下同。
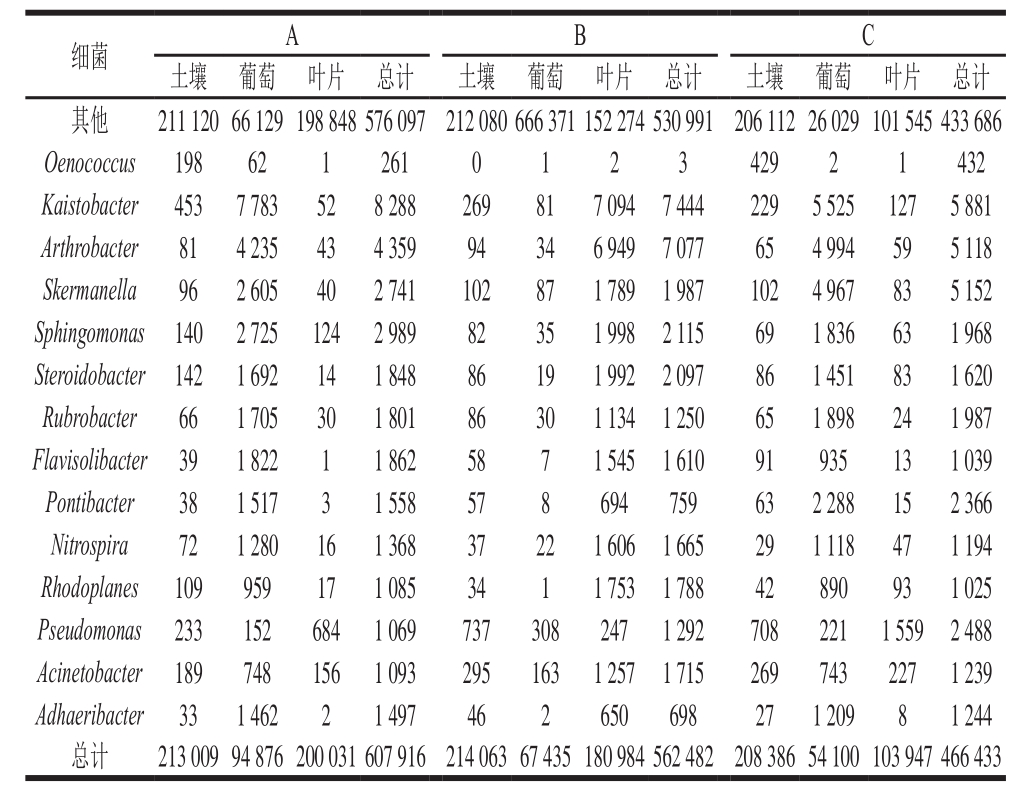
表2显示了样品的真菌群落构成及大小,从不同的葡萄园来看,B地区检测出的真菌数量最多,其次为C地区、A地区;从样品种类来看,土壤所检测出的真菌数量是最高的,其次是葡萄叶片,最后是葡萄表面。表3显示了样品的细菌群落构成及大小,从不同的葡萄园来看,A地区检测出的细菌数量最多,其次为B地区、C地区;从样品种类来看,同真菌检测结果相同,土壤所检测出的细菌数量是最高的,其次是葡萄叶片,最后是葡萄表面。
表3 3 个不同地区土壤、葡萄和叶片中属水平细菌微生物数量
Table 3 Bacterial quantities at family level in soil, as well as on grapes and leaves
2.2 微生物丰度分析
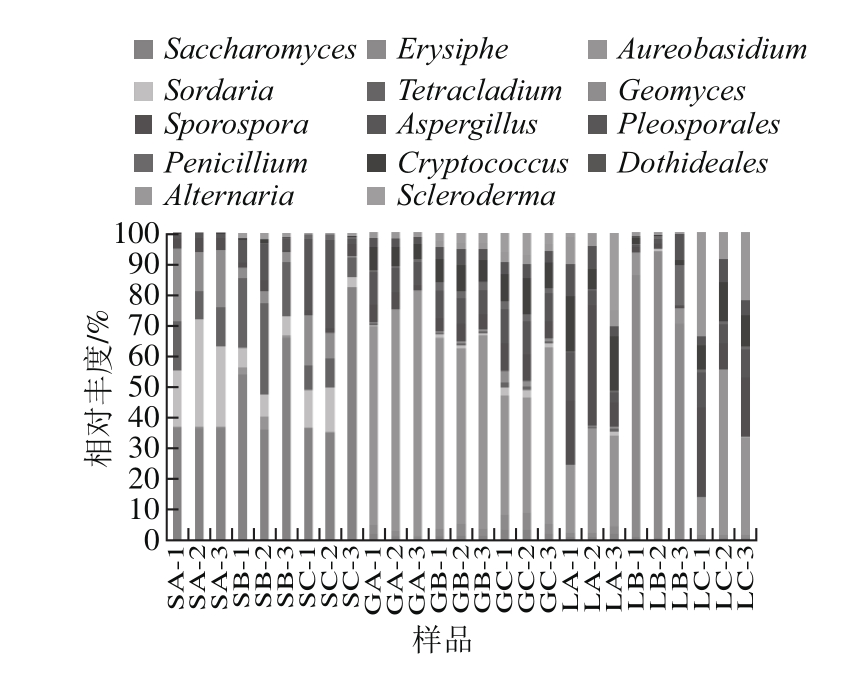
图1 真菌群落属水平相对丰度
Fig. 1 Relative abundance of fungi at genus level
由于样品中所检测出的微生物种类繁多,许多物种含量十分少,不能明显地与含量多的物种表示在同一个图中,所以以物种的丰度大于1%作为划分依据,分别选择了排名前14的真菌、细菌优势菌属,以相对丰度为纵坐标,绘制柱形图。图1显示出相对丰度值高于1%的14 种真菌菌属,其中,Erysiphe在A、C地区土壤中含量很低甚至未检出,Sordaria和Tetracladium在A地区葡萄叶片中含量很低甚至未检出,Tetracladium在A地区葡萄、Geomyces在C地区叶片中含量很低甚至未检出。在土壤样品中Saccharomyces最为丰富,其次是Sordaria、Tetracladium和Geomyces;在葡萄中,Aureobasidium是最丰富的,其次为Cryptococcus、Aspergillus和Pleosporales;葡萄叶片中最丰富的也是Aureobasidium,其次为Sporospora。
Saccharomyces、Aureobasidium、Sporospora和Aspergillus虽然丰度有所不同,但出现在所有样品中。Saccharomyces、Sordaria和Tetracladium大量存在于土壤样品中,而在葡萄表面尤其是叶片中含量十分少。Aureobasidium大量存在于葡萄表面,其次为叶片,在土壤中含量却十分少。Pleosporales、Penicillium、Cryptococcus、Dothideales、Alternaria和Scleroderma也出现在所有样品中,但丰度较小。值得注意的是,Erysiphe在B地区葡萄叶片中大量存在,几乎接近于菌属总和的50%。

图2 细菌群落属水平相对丰度
Fig. 2 Relative abundance of bacteria at genus level
图2 显示,在27组样品中,其他未检出菌属所占比例较大,只有Arthrobacter、Sphingomonas、Steroidobacter、Pseudomonas和Acinetobacter 5 种细菌菌属存在于所有土壤、叶片和葡萄中,其他菌属尤其是Oenococcus含量很低甚至未检出。土壤中细菌检出量明显大于叶片,葡萄表面所检出的细菌最少。
土壤中丰度最大的菌属为Kaistobacter,其次为Arthrobacter、Skermanella;葡萄和叶片中Pseudomonas丰度相对较大,其次为Sphingomonas和Adhaeribacter。Flavisolibacter在A地区葡萄中、Adhaeribacter在B地区葡萄中未检出。所有样品中虽然检出有醋酸杆菌,但其含量均很低,没有进入前14 种优势菌属范围。
2.3 样品中所含OTU数目分析


图3 真菌群落维恩图
Fig. 3 Venn diagram of fungal communities
采用Qiime软件,得到每个OTU的分类学信息,通过R软件,绘制维恩图。由图3a看出,3 个葡萄园的土壤样品中分别含有真菌种类150、156、126、141、142、137、131、146 个和143 个,其中共有的菌种种类有94 个;由图3b看出,葡萄果实样品中分别含有真菌种类91、98、80、118、104、103、97、110 个和106 个,共有的真菌种类有69 个;由图3c看出,葡萄叶片样品中分别含有真菌种类79、80、77、99、73、90、87、101 个和89 个,共有的真菌种类58 个。3 个地区葡萄园土壤中所含的相同真菌种类较多,而葡萄叶片中较少。
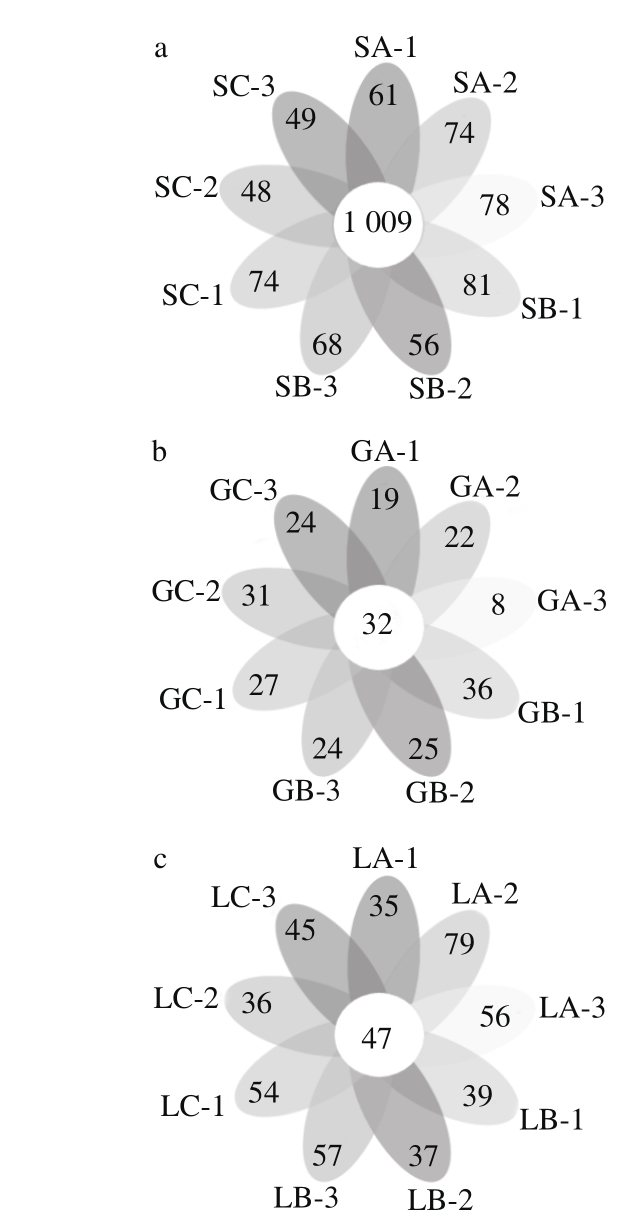
图4 细菌群落维恩图
Fig. 4 Venn diagram of bacterial communities
由图4a可看出,3个葡萄园的土壤样品中分别含有细菌种类1 070、1 083、1 087、1 090、1 065、1 077、1 083、1 057个和1 058 个,共有的细菌种类1 009 个;由图4b看出,葡萄样品中分别含有的细菌种类51、54、40、68、57、56、59、63 个和56 个,有32 种细菌在所有葡萄样品中均含有;由图4c看出,葡萄叶片中分别含有细菌种类82、126、103、86、84、104、101、83 个和92 个,共有的细菌种类47 个。从细菌OTU数目分析,不同地区土壤中相同微生物比较多,而葡萄叶片和葡萄果实相对较少。
2.4 不同样品中微生物群落的比较分析
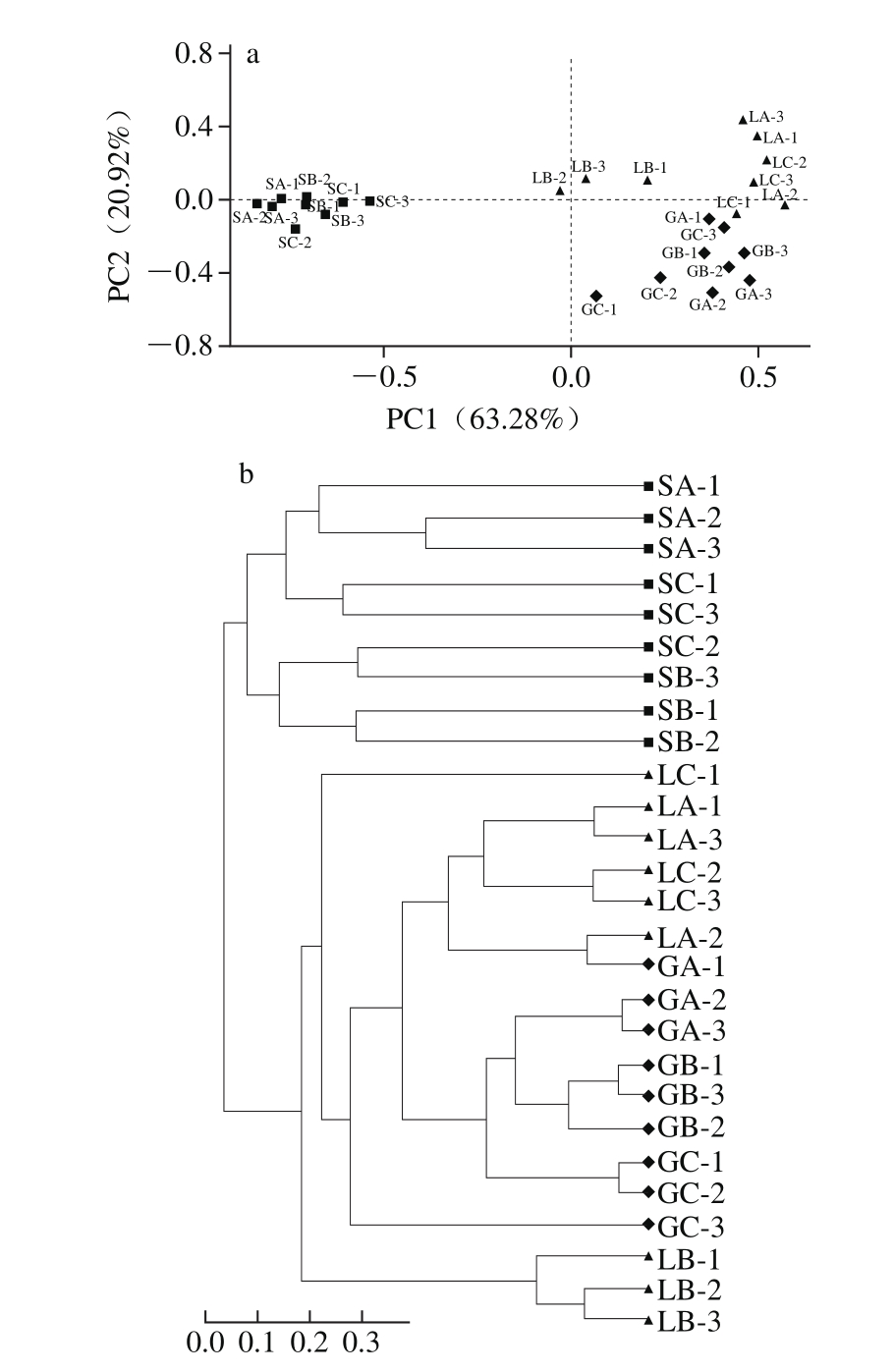
图5 真菌群落主成分分析(a)和UPGMA聚类分析(b)图
Fig. 5 Principal coordinate analysis of fungal communities (a) and UPGMA cluster analysis (b) of the fungal microbiota
用R语言分析3 个葡萄园样品的真菌群落,不同样品分别用不同的点来表示,进行主成分分析及聚类分析。图5a中,当PC1为63.28%,PC2为20.92%时,从样品种类来看,葡萄叶片和葡萄果实中的真菌群落相似性较高,相比之下,土壤与叶片、葡萄的距离较远,相似性较低。从样品采集地点来看,3 个葡萄园中土壤的相似性较高,B葡萄园中的叶片微生物与A、C地区有清晰的划分,而A、C地区的酿酒葡萄虽然与B地区之间有一定的距离,但差异较小。
为了比较不同地区样品间微生物种类的差异,本研究将所有样品真菌群落进行聚类分析(图5b),结果表明,在遗传距离为0.05处时,土壤样品与葡萄叶片、酿酒葡萄产生了分支,同时,3 个葡萄园中的土壤也各成一个分支,差异明显。葡萄叶片与葡萄差异较小,B葡萄园的葡萄叶片在0.1遗传距离处与A、C葡萄园产生分支,各葡萄园中的葡萄无明显差异。
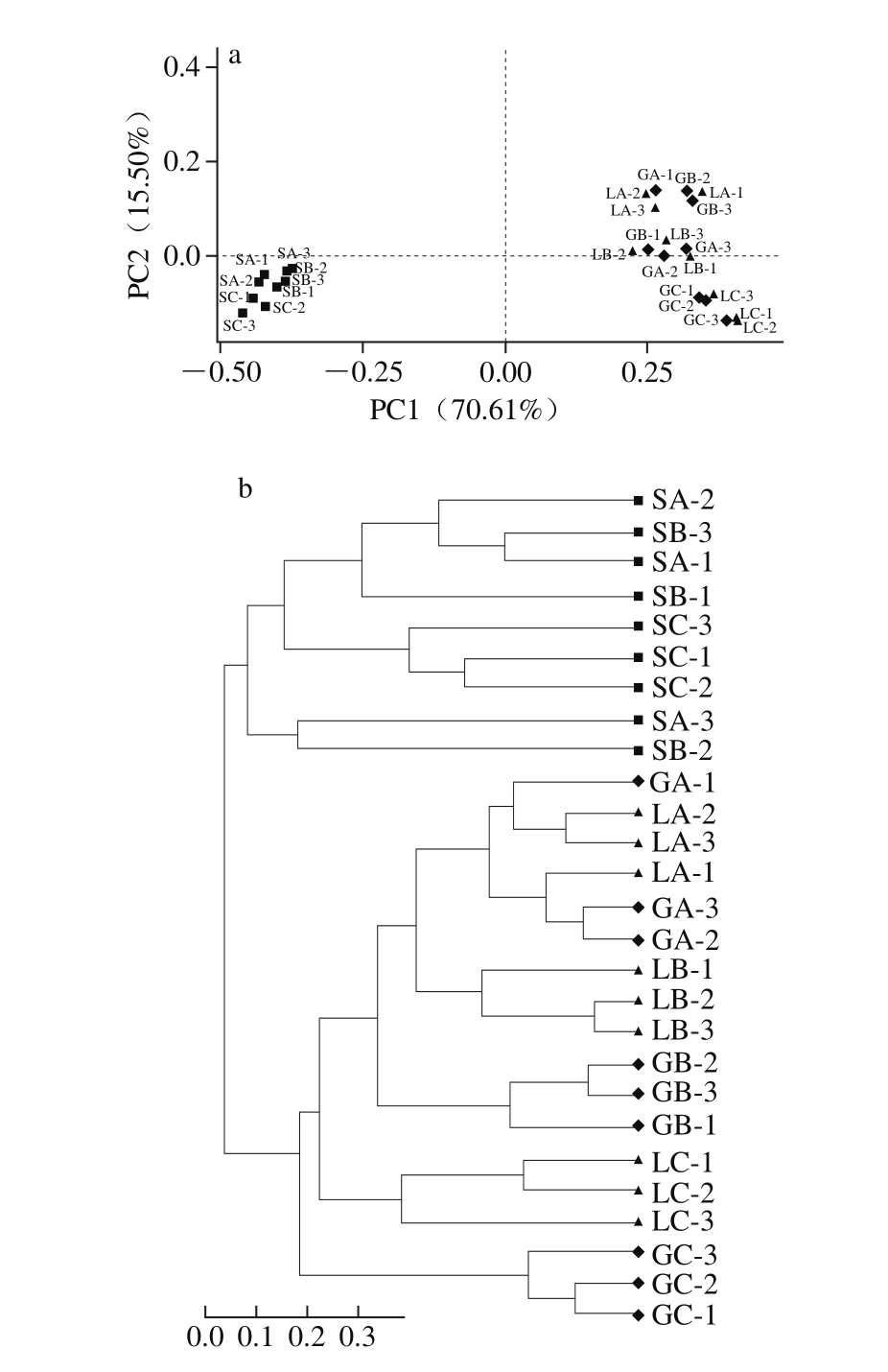
图6 细菌群落主成分分析(a)和UPGMA聚类分析(b)图
Fig. 6 Principal coordinate analysis of bacterial communities (a) and UPGMA cluster analysis (b) of the bacterial microbiota
与真菌群落相同,用SPSS分析3 个葡萄园样品的细菌群落。图6a显示,当PC1为70.61%,PC2为15.50%时,从样品种类来看,葡萄叶片和葡萄中的真菌群落相似性十分高,相比之下,土壤与叶片、葡萄的距离较远,相似性较低。从样品采集地点来看,3 个葡萄园中土壤的相似性较高,其中A和B葡萄园更为相似,C葡萄园中的微生物与A、B地区存在差异;A、B葡萄园的叶片和葡萄聚集在一起,无明显的差异,而C葡萄园相对于A、B葡萄园来说,有明显的区别,存在差异。
将所有样品的细菌群落进行聚类分析(图6b),结果与主成分分析结果基本一致。在遗传距离为0.05处时,所有的土壤样品就与葡萄叶片、葡萄产生了分支,但3 个葡萄园中土壤的划分不是十分清晰,C与A、B葡萄园不在同一条分支上,而A和B葡萄园交互在一起,无明显差异。B、C葡萄园中的叶片和葡萄均各自产生一条分支,有明确的区别,只有A葡萄园中的叶片和葡萄无明显差异。
3 讨 论
近年来,对于葡萄酒微生物的研究已不只局限于酿酒葡萄浆果,而是逐渐扩大到了葡萄叶片、葡萄树皮、葡萄园土壤乃至更大的生态系统,许多研究都表明与葡萄酒相关的真菌和细菌微生物会附生在葡萄树的各个部位上,如葡萄汁、葡萄酒、葡萄叶片、树皮、树根、土壤等[24-26]。
Renouf等[27]研究表明,土壤中的优势真菌菌属为Saccharomyces、Sordaria、Tetracladium和Geomyces,葡萄和葡萄叶片中的优势真菌菌属为Aureobasidium、Sporospora、Cryptococcus和Dothideales,此外,本研究还检测到了其他真菌菌属如Aspergillus、Pleosporales、Penicillium、Erysisphe、Alternaria和Scleroderma;Pinto[28]和Stevanato[29]等研究表明,Ascomycota和Basidiomycot是土壤、葡萄和葡萄叶片中主要的真菌微生物(门水平),本研究通过检测不仅得到了Ascomycota和Basidiomycot,还得到了Chytridiomycota,Un—s-fungal sp CC 06_28和Zygomycota。需要注意的是,已有研究[30-32]指出,葡萄树白粉病对葡萄伤害非常大,使葡萄幼果易开裂腐败,该病由Erysisphe引发,但目前可以有效预防,本研究在B地区的葡萄叶片上检测出了大量的Erysisphe,建议B地区需要尽快进行防治;已有研究[33-35]指出,赭曲霉素可以致人死亡,它主要由Aspergillus产生,是全球葡萄酒都存在的问题,有些权威机构建立了2 μg/L的最大限量,虽然如此,但它存在于葡萄和葡萄酒中的可能性风险却不高;Lachenmeier等[36]指出,霉菌是一种丝状真菌,大量存在于葡萄表面,能够侵染葡萄的茎、叶、花、果实等,造成果实减产,影响果实品质,它在储酒橡木桶内外、葡萄酒瓶木塞处很容易生长繁殖,当浓度达到十亿分之一或万亿分之一时就能明显的闻出,产生令人不愉快的霉味,部分霉菌的次级代谢产物还有致癌趋势,损害人体健康。
Hirano[37]、Park[38]等研究表明,土壤中的优势细菌菌属为Kaistobacter、Arthrobacter、Skermanella和Sphingomonas,葡萄和葡萄叶片中的优势细菌菌属为Pseudomonas、Acinetobacter和Kaistobacter,此外,本研究还检测到了其他细菌菌属如Steroidobacter、Rubrobacter、Flavisolibacter、Pontibacter、Nitrospira、Rhodoplanes和Adhaeribacter;Proteobacteria、Firmicutes、Bacteroidetes和Actinobacteria为土壤、葡萄和葡萄叶片中主要的细菌微生物(门水平),本研究通过检测不仅得到了Proteobacteria、Firmicutes、Bacteroidetes和Actinobacteria,还得到了Crenarchaeota、Nitrospirae、Planctomycetes和Verrucomicrobia;李翠霞[39]通过对Oenococcus的分离鉴定得出,Oenococcus在葡萄酒乳酸发酵阶段数量迅速增长成为优势菌属,并大量存在于葡萄酒中,本研究也证明了Oenococcus并非土壤、葡萄、葡萄叶片中的优势菌。
众所周知,酵母菌是葡萄酒酿造的灵魂,直接决定了葡萄酒的风味及感官品质。目前葡萄酒相关酵母菌种类繁多,分属于24 个属的120 种,几乎遍及酵母菌的主要属种,主要有Saccharomyces、Pichia、Candida、Hanseniaspora、Schizosaccharomyces、Dekkera、Metschnikowia、Issatchenkia、Saccharomycodes、Zygosaccharomyces 10 个属的酵母,其中最具代表性的为酵母属。本研究检测到的Saccharomyces,可以将葡萄浆果中的糖转化成乙醇、CO2及其他代谢产物,在葡萄汁发酵过程中生成并释放多种香气物质,同时还对葡萄果实表面的细菌有一定的生物抑制作用[40-41],一些非酿酒酵母经过恰当利用控制,还可有效提高葡萄酒风味及复杂性[42-44]。在真菌微生物中,受贵腐霉菌侵染的葡萄所酿造的贵腐葡萄酒,风味与其他葡萄酒迥然不同,口味独特,是一种少见的高档酒[45]。
与真菌相比,除了部分乳酸菌外,大多数细菌微生物对于葡萄酒来说都是有害菌,会在不同程度上对葡萄酒的品质造成影响,例如检测到的Proteobacteria,醋酸菌是葡萄酒酿造中典型的有害菌[46],它能够将乙醇转化成乙酸,导致葡萄酒酸败甚至变质。因此,在酿造过程的各个阶段都应该尽可能避免或减少细菌侵染,并且跟踪监测葡萄酒中的微生物状况,以确保葡萄酒的良好品质。葡萄酒相关细菌主要为乳酸菌和醋酸菌,乳酸菌有酒球菌属(Oenococcus)、乳杆菌属(Latobacillus)、明串珠菌属(Leuconostoc)、片球菌属(Pediococcus),其中酒类酒球菌(Oenococcusoeni)是进行苹果酸-乳酸发酵(malate fermentation,MLF)的主要菌种,部分短乳杆菌、植物乳杆菌也可以引发苹乳发酵[47-48],本研究检测到的Firmicutes,发生MLF后可以有效的降低葡萄酒酸度,避免葡萄酒在装瓶、贮存期间发生二次发酵,增加葡萄酒的微生物学稳定性,乳酸菌的代谢可以改变葡萄酒中醛类、酯类等呈香物质和氨基酸、有机酸、维生素等微量元素的成分及含量,从而影响葡萄酒的感官品质[49-50]。
传统培养方法结合PCR如限制片段长度多态性分析、随机扩增多态性分析技术及一些非培养方法如变性梯度凝胶电泳、实时荧光PCR技术和荧光原位杂交技术存在费力、耗时、结果可靠性差等缺陷[51-53],本研究利用高通量测序技术通量高、产生数据量大、测序周期短、准确率高以及成本低等在微生物多样性研究中具有的优越性,将该技术运用于葡萄酒微生物多样性分析,研究表明,良好的环境有利于大多数微生物的生长,影响着土壤、葡萄、葡萄叶片中微生物群落多样性。从样品种类来说,土壤比葡萄、葡萄叶片中的真菌和细菌种类多、数量多,早期研究表明,葡萄园土壤可以说是一个天然的微生物菌库[54],它提供着微生物生长所需要的营养,如碳源(氨基酸、有机酸和碳水化合物)、氮源和生长因子等[55],其本身的理化性质如质地、矿物成分、有机质等,直接影响着微生物的生长和分布,而施肥、耕作、种植等不同葡萄园管理方式及重金属、有机污染物、工厂废弃物等外部因素又可能会影响土壤的物理化学性质,从而改变微生物群落的组成和分布[56-62]。从样品所在地区来说,A、B地区冬季严寒、干燥,夏季酷热,C地区气候条件相对较温和,冬季寒冷,夏季炎热,3 个葡萄园日照充裕,昼夜温差大,降水量少,病虫害少,均为种植酿酒葡萄的优质产区。但C地区微生物多样性低于A、B地区,其原因主要在于C地区的气候条件,春季及早夏的大风、沙尘天气,严重影响着微生物的生长[63-64]。
本研究通过检测分析新疆A、B、C 3 个地区葡萄园土壤、葡萄和葡萄叶片中的微生物种类、数量及分布,对新疆地区葡萄酒相关微生物群落多样性有了一定的了解,为揭示新疆地区葡萄酒相关微生物多样性、建立葡萄酒微生物种质资源库、筛选优良品种、提高葡萄酒品质等方面提供了理论支持。
参考文献:
[1] PRAKICHAIWATTANA C J, FLEET G H, HEARD G M. Application and evaluation of denaturing gradient gel electrophoresis to analyse the yeast ecology of wine grapes[J]. FEMS Yeast Research, 2004, 4(8):865-877. DOI:10.1016/j.femsyr.2004.05.004.
[2] RENOUF V, CLAISSE O, LONVAUD-FUNEL A. Inventory and monitoring of wine microbial consortia[J]. Applied Microbiology and Biotechnology, 2007, 75(1): 149-164. DOI:10.1007/s00253-006-0798-3.
[3] OCÓN E, GUTIERREZ A R, GARIJO P, et al. Presence of non-Saccharomyces yeasts in cellar equipment and grape juice during harvest time[J]. Food Microbiology, 2010, 27(8): 1023-1027.DOI:10.1016/j.fm.2010.06.012.
[4] JOLLY N P, VARELA C, PRETORIUS I S. Not your ordinary yeast:non-Saccharomyces yeasts in wine production uncovered[J]. FEMS Yeast Research, 2014, 14(2): 215-237. DOI:10.1111/1567-1364.12111.
[5] RIBEREAU-GAYON P, DUBOURDIEU D, DONECHE B. Handbook of enology: the microbiology of wine and vinifications[M]. New York:John Wiley and Sons, 2006: 18-37. DOI:10.1002/0470010363.ch1.
[6] SUN H H, MA H Q, HAO M L, et al. Identification of yeast population dynamics of spontaneous fermentation in Beijing wine region, China[J].Annals of Microbiology, 2009, 59(1): 69-76. DOI:10.1007/BF03175601.
[7] LI E, LIU A, XUE B, et al. Yeast species associated with spontaneous wine fermentation of Cabernet Sauvignon from Ningxia, China[J].World Journal of Microbiology & Biotechnology, 2011, 27(10): 2475-2482. DOI:10.1007/s11274-011-0711-9.
[8] 王慧, 张立强, 刘天明, 等. 葡萄果粒表皮酵母菌多样性研究[J]. 微生物学通报, 2008, 35(1): 10-14. DOI:10.3969/j.issn.0253-2654.2008.01.004.
[9] 徐艳文, 刘爱国, 刘延琳, 等. 甘肃莫高葡萄酒厂酵母种群的生态分布[J]. 生态学报, 2009, 29(6): 328-333. DOI:10.3321/j.issn:1000-0933.2009.06.039.
[10] 李桥. 基于高通量测序技术下土壤微生物群落结构的研究[D]. 济南:山东师范大学, 2014: 22-45.
[11] BOKULICH N A, THORNGATE J H, RICHARDSON P M, et al.Microbial biogeography of wine grapes is conditioned by cultivar,vintage, and climate[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(1): 139-148.DOI:10.1073/pnas.1317377110.
[12] 宋培勇, 马莉莉. TENP-PBS预处理在提取土壤DNA中的应用[J].安徽农业学报, 2010, 38(24): 12978-12980. DOI:10.3969/j.issn.0517-6611.2010.24.019.
[13] 赵怀宝, 冯建荣, 蒋迪军, 等. 葡萄DNA提取与纯化方法的比较[J].石河子大学学报(自然科学版), 2000, 4(2): 117-121. DOI:10.3969/j.issn.1007-7383.2000.02.007.
[14] 遆卫国. 葡萄不同部位提取DNA的方法研究[J]. 山西师范大学学报(自然科学版), 2002, 16(1): 63-65. DOI:10.3969/j.issn.1009-4490.2002.01.015.
[15] ZHAN B, FADISTA J, THOMSEN B, et al. Global assessment of genomic variation in cattle by genome resequencing and highthroughput genotyping[J]. BMC Genomics, 2011, 12(2): 557.DOI:10.1186/1471-2164-12-557.
[16] 许宇静, 张煜坤, 沈雪梅, 等. 采用Illumina MiSeq测序技术分析断奶幼兔盲肠微生物群落的多样性[J]. 动物营养学报, 2015, 27(9):2793-2802. DOI:10.3969/j.issn.1006-267x.2015.09.017.
[17] COLE J R, WANG Q, FISH J A, et al. Ribosomal Database Project:data and tools for highthroughput rRNA analysis[J]. Nucleic Acids Research, 2014, 42(D1): 633-642. DOI:10.1093/nar/gkt1244.
[18] MOSHER J J, BERNBERG E L, SHEVCHENKO O, et al. Eきcacy of a 3rd generation high-throughput sequencingplatform for analyses of 16S rRNA genes from environmental samples[J]. Journal of Microbiological Methods, 2013, 95(2): 175-181. DOI:10.1016/j.mimet.2013.08.009.
[19] AMNON A, AMIT Z, OR Z, et al. High-resolution microbial community reconstruction byintegrating short reads from multiple 16S rRNA regions[J]. Nucleic Acids Research, 2013, 41(22): 205-219.DOI:10.1093/nar/gkt1070.
[20] SHIRAYAMA M, SETH M, LEE H, et al. Supplementary material-search and clustering orders of magnitude faster than BLAST[J].Nucleic Acids Research, 2012, 26(1): 1-28. DOI:10.1038/nmeth0809-550.
[21] CAPORASO J G, BITTINGER K, BUSHMAN F D, et al. PyNAST: a flexible tool for aligning sequences to atemplate alignment[J]. Bioinformatics,2010, 26(2): 266-267. DOI:10.1093/bioinformatics/btp636.
[22] CAPORASO J, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows integration and analysis of high-throughput community sequencing data. Nat. Meth[J]. Nature Methods, 2010, 7(5): 335-336.
[23] MCCAIN A E, GLOVER L A, PROSSER J I. Molecular analysis of bacterial community structure and diversity in unimproved and improved upland grass pastures[J]. Applied and Environmental Microbiology, 1999, 65(4): 1721-1730.
[24] MARTINS G, MIOT-SERTIER C, LAUGA B. Grape berry bacterial microbiota: impact of the ripening process and the farming system[J].international Journal of Food Microbiology, 2012, 158(2): 93-100.DOI:10.1016/j.ijfoodmicro.2012.06.013.
[25] VERGINER M, LEITNER E, BERG G. Production of volatile metabolites by grape-associated microorganisms[J]. Journal of Agricultural and Food Chemistry, 2010, 58(14): 8344-8350.DOI:10.1021/jf100393w.
[26] BOULTON R B, SINGLETON V L, BISSON L F, et al. Principles and practices of wine making[M]. New York: Springer US, 1999: 535-542.DOI:10.1007/978-1-4757-6255-6_1.
[27] VINCENT R, OLIVIER C, ALINE L. Understangding tne microbial ecosystem on the grape herry surfur through numeration and identification of yeast and bacteria[J]. Australian Journal of Grape and Wine Research, 2010, 11(3): 316-327. DOI:10.1111/j.1755-0238.2005.tb00031.x.
[28] PINTO C, PINHO D, SOUSA S, et al. Unravelling the diversity of grapevine microbiome[J]. PLoS ONE, 2014, 9(1): 85622.DOI:10.1371/journal.pone.0085622.
[29] STEVANATO P, BERTAGGIA M, STELLIN F. Soil biological and biochemical traits linked to nutritional status in grapevine[J].Journal of Soil Science and Plant Nutrition, 2014, 14(3): 469-479.DOI:10.4067/S0718-95162014005000033.
[30] HAN L J, WENG K, MA H, et al. Identification and characterization of Erysiphe necator-responsive microRNAs in Chinese wild Vitis pseudoreticulata by high-throughput sequencing[J]. Frontiers in Plant Science, 2016, 7: 621. DOI:10.3389/fpls.2016.00621.
[31] GAAO Y R, HAN Y T, ZHAO F L, et al. Identification and utilization of a new Erysiphe necator isolate NAFU1 to quickly evaluate powdery mildew resistance in wild Chinese grapevine species using detached leaves[J]. Plant Physiology and Biochemistry, 2016, 98: 12-24.DOI:10.1016/j.plaphy.2015.11.003.
[32] AMRINE K C, BLANCO-ULATE B, RIAZ S, et al. Comparative transcriptomics of central Asian Vitis vinifera accessions reveal distinct defense strategies against powdery mildew[J]. Horticulture Research,2015, 2: 15037. DOI:10.1038/hortres.2015.37.
[33] GARMENDIA G, VERO S. Occurrence and biodiversity of Aspergillus section Nigri on ‘Tannat’ grapes in Uruguay[J].International Journal of Food Microbiology, 2016, 216: 31-39.DOI:10.1016/j.ijfoodmicro.2015.08.020.
[34] FERRARI V, DELLACASSA E, CONIBERTI A, et al. Role of grapevine vegetative expression on Aspergillus spp. incidence and OTA accumulation in wines produced in a temperate humid climate[J].Food Additives & Contaminants: Part A: Chemistry, Analysis, Control,Exposure & Risk Assessment, 2017, 34(2): 299-306. DOI:10.1080/194 40049.2016.1252064.
[35] QI T F, RENAUD J B, MCDOWELL T, et al. Diversity of mycotoxinproducing black Aspergilli in Canadian vineyards[J]. Journal of Agricultural and Food Chemistry, 2016, 64(7): 1583-1589.DOI:10.1021/acs.jafc.5b05584.
[36] LACHENMEIER D W, KANTERES F, KUBALLA T, et al. Ethyl carbamate in alcoholic beverages from Mexio (tequila, mescal,stool) and Guatemala (cuxa): market survey and risk assessment[J].International Journal of Environmental Research and Public Health,2009, 6(1): 349-360. DOI:10.3390/ijerph6010349.
[37] HIRANO S S, UPPER C D. Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae-a pathogen, ice nucleus, and epiphyte[J]. Microbiology and Molecular Biology Reviews, 2000,64(3): 624-653. DOI:10.1128/MMBR.64.3.624-653.2000.
[38] PARK J Y, HAN S H, LEE J H, et al. Draft genome sequence of the biocontrol bacterium Pseudomonas putida B001, an oligotrophic bacterium that induces systemic resistance to plant diseases[J]. Journal of Bacteriology, 2011, 193(23): 6795-6796. DOI:10.1128/JB.06217-11.
[39] 李翠霞. 中国优良酒类酒球菌的分离、鉴定[D]. 杨凌: 西北农林科技大学, 2011: 28-35.
[40] 刘爱国. 宁夏贺兰山麓葡萄酿酒酵母菌的分离及其分类鉴定[D].杨凌: 西北农林科技大学, 2008: 24-40.
[41] FUELSANG K C, EDWARDS C G. Wine microbio practical applications and procedures[M]. New York: Springer US, 2007: 44-52. DOI:10.1007/978-0-387-33349-6.
[42] 韩珊珊. 柠檬形克勒克酵母(Kloeckera apiculata)在葡萄酒发酵中的应用研究[D]. 杨凌: 西北农林科技大学, 2008: 19-32.
[43] 徐亚男, 李琦, 刘秋萍. 新疆葡萄酒产区非酿酒酵母菌研究进展[J].食品工业, 2015, 36(1): 266-270.
[44] HONG Y A, PARK H D. Role of non-Saccharomyces yeasts in Korean wines produced from campbell early grapes: potential use of Hanseniaspora uvarum as a starter culture[J]. Food Microbiology,2013, 34(1): 207-214. DOI:10.1016/j.fm.2012.12.011.
[45] 张春芝, 江志国. 微生物对葡萄酒香气的影响综述[J]. 中国酿造,2013, 32(9): 28-31. DOI:10.3969/j.issn.0254-5071.2013.09.008.
[46] 屈慧鸽. 葡萄酒生产过程中醋酸菌的危害及影响因素分析[J]. 酿酒科技, 2009(2): 43-46. DOI:10.13746/j.njkj.2009.02.030.
[47] MAICAS S. The use of alternative technologies to develop malolactic fermentation in wine[J]. Applied Microbiology and Biotechnology,2001, 56(1): 35-39. DOI:10.1007/s002530100662.
[48] 张春晖, 李华. 葡萄酒微生物学[M]. 西安: 陕西人民出版社, 2003:1-7.
[49] 周利国. 宁夏银川葡萄酒产区酒酒球菌分离筛选及鉴定[D]. 杨凌:西北农林科技大学, 2009: 22-38.
[50] BLATTEL V, WIRTH K, CLAUS H, et al. A lytic enzyme cocktail from Streptomyces sp. B578 for the control of lactic and acetic acid bacteria in wine[J]. Applied Microbiology and Biotechnology, 2009,83(5): 839-848. DOI:10.1007/s00253-009-1926-7.
[51] COCOLIN L, BISSON L F, MILLS D A. Direct profiling of the yeast dynamics in wine fermentations[J]. FEMS Microbiology Letters, 2000,189(1): 81-87. DOI:10.1111/j.1574-6968.2000.tb09210.x.
[52] PRAKICHAIWATTANA C J, FLEET G H, HEARD G M.Application and evaluation of denaturing gradient gel electrophoresis to analyse the yeast ecology of wine grapes[J]. FEMS Yeast Research,2004, 4(8): 865-877. DOI:10.1016/j.femsyr.2004.05.004.
[53] RENOUF V, CLAISSE O, LONVAUA-FUNEL A. Inventory and monitoring of wine microbial consortia[J]. Applied Microbiology and Biotechnology, 2007, 75(1): 149-164. DOI:10.1007/s00253-006-0798-3.
[54] MARTINS G, LAUGA B, MIOT-SERTIRE C, et al. Characterization of epiphytic bacterial communities from grapes, leaves, bark and soil of grapevine plants grown, and their relations[J]. PLoS ONE, 2013,8(8): 73013. DOI:10.1371/journal.pone.0073013.
[55] WAWRIK B, KERKHOF L, KUKOR J, et al. Effect of different carbon sources on community composition of bacterial enrichments from soil[J]. Applied and Environmental Microbiology, 2005, 71(11):6776-6783. DOI:10.1128/AEM.71.11.6776-6783.2005.
[56] GANS J, WOLINSKY M, DUNBAR J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil[J].Science, 2005, 309: 1387-1390. DOI:10.1126/science.1112665.
[57] DELL’AMICO E, MAZZOCCHI M, CAVALCA L, et al. Assessment of bacterial community structure in a long-term copper-polluted exvineyard soil[J]. Microbiological Research, 2008, 163(6): 671-683.DOI:10.1016/j.micres.2006.09.003.
[58] SESSITSCH A, WEILHARTER A, GERZABEK M H, et al. Microbial population structures in soil particle size fractions of a long-term fertilizer field experiment[J]. Applied and Environmental Microbiology, 2001,67(9): 4215-4224. DOI:10.1128/AEM.67.9.4215-4224.2001.
[59] KAMMA M, MBURU H, BLANCHART E, et al. Effects of organic and inorganic fertilization on soil bacterial and fungal microbial diversity in the Kabete long-term trial, Kenya[J]. Biology and Fertility of Soils, 2011, 47(3): 315-321. DOI:10.1007/s00374-011-0539-3.
[60] TAN Y, CUI Y, LI H, et al. Rhizospheric soil and root endogenous fungal diversity and composition in response to continuous Panax notoginseng cropping practices[J]. Applied and Environmental Microbiology, 2017, 194: 10-19. DOI:10.1016/j.micres.2016.09.009.
[61] CORDERO-BUESO G, ARROYO T, SERRANO A, et al. Influence of different floor management strategies of the vineyard on the natural yeast population associated with grape berries[J]. International Journal of Food Microbiology, 2011, 148(1): 23-29. DOI:10.1016/j.ijfoodmicro.2011.04.021.
[62] CORDERO-BUESO G, ARROYO T, SERRANO A, et al. Influence of the farming system and vine variety on yeast communities associated with grape berries[J]. International Journal of Food Microbiology,2011, 145(1): 132-139. DOI:10.1016/j.ijfoodmicro.2010.11.040.
[63] 孙殿明, 赵萍, 孙颜琪. C地区县酿酒葡萄产业发展的农业气候条件分析[J]. 农业与技术, 2015, 35(17): 149-151. DOI:10.11974/nyyjs.20150932058.
[64] 常绪正, 康永义. 222团发展酿酒葡萄的气候条件分析[J]. 新疆气象,2001, 24(5): 27-28. DOI:10.3969/j.issn.1002-0799.2001.05.009.