类固醇激素是一类具有强且持久的蛋白同化作用的四环脂肪烃化合物,包括天然及人工合成的糖皮质激素、孕激素、雌激素等。由于类固醇激素具有较强的抗炎作用,并可促进动物超常态生长,能够大幅提高动物养殖经济效益,在养殖业中经常被大量使用。滥用该类激素则会造成动物乳源不同程度药物残留,人类食用后可导致机体代谢紊乱、发育异常或潜在致癌、致畸风险[1]。因此世界各国均规定了动物源性食品中各种激素的最大残留量,欧盟第96/22/EC指令、美国食品药品管理局、日本肯定列表也禁止在食品源性动物中使用激素类药物。我国农业部文件《动物性食品中兽药最高残留限量》[2]规定牛奶中氢化可的松的最大残留量为10 μg/kg,欧盟规定地塞米松、泼尼松龙在牛奶中的最大残留量分别为0.3、6 μg/kg[3]。为加强对乳制品污染的监控,有效监督我国奶粉质量现状,必须发展灵敏可靠的多种激素检测方法,以期通过有效监测,对可能发生的污染状况进行预测,并采取必要的措施预防食品危害的发生。
牛奶和奶粉中除了含有内源性孕激素、雌激素外,其他所有化学合成类激素均属于禁用药物。目前国内外已有许多激素检测的标准与文献,但对乳制品中多激素残留检测方法却少有报道,国家标准暂未发布权威的检测方法。检测类固醇激素主要有气相色谱法[4]、高效液相色谱法[5-8]、气相色谱-质谱法[9-12]和液相色谱-质谱法[13-17]。气相色谱和高效液相色谱法方法灵敏度较低,选择性和特异性差,不适合多激素药物残留的分析。气相色谱-质谱方法虽然灵敏度和特异性都很高,可以满足残留分析的要求,但衍生过程繁琐。高效液相色谱-串联质谱是分析动物源性食品中激素残留的一种重要方法,该方法灵敏度高,选择性和特异性好,能够对低浓度样品很好地确认,已用于多类复杂基质中的激素测定[18-20]。
Anastassiadas等2003年开发了分散固相萃取技术QuEChERS(quick, easy, cheap, effective, rugged and safe)[21],这是一种将目标化合物的提取或分离与净化相结合的原创方法,最早在农药多残留中使用[22-25]。本实验将QuEChERS技术应用于样品前处理,结合超高效液相色谱-串联质谱,建立牛奶和奶粉中42 种类固醇激素残留量的检测方法,该方法简单、快速、准确。
1 材料与方法
1.1 材料与试剂
市售样品牛奶、奶粉,均为监督抽检试样。
42 种类固醇激素标准品:泼尼松龙(纯度99.4%)、氢化可的松(纯度99.8%)、泼尼松(纯度99.7%)、可的松(纯度98%)、甲基泼尼松龙(纯度99.5%)、倍他米松(纯度98.8%)、地塞米松(纯度98.9%)、氟米松(纯度98.0%)、倍氯米松(纯度96.0%)、曲安奈德(纯度98.4%)、氟氢缩松(纯度99.4%)、曲安西龙双醋酸酯(纯度98.9%)、泼尼松龙醋酸酯(纯度99.1%)、氟米龙(纯度99.6%)、氢化可的松醋酸酯(纯度99.5%)、地夫可特(纯度98.0%)、氟氢可的松醋酸酯(纯度98%)、泼尼松醋酸酯(纯度99.5%)、可的松醋酸酯(纯度99.4%)、甲基泼尼松龙醋酸酯(纯度99.1%)、倍他米松醋酸酯(纯度97.5%)、布地奈德(纯度98.0%)、氢化可的松丁酸酯(纯度99.5%)、地塞米松醋酸酯(纯度98.0%)、氟米龙醋酸酯(纯度99.2%)、氢化可的松戊酸酯(纯度99.5%)、曲安奈德醋酸酯(纯度99.1%)、氟轻松醋酸酯(纯度98%)、二氟拉松双醋酸酯(纯度98.6%)、倍他米松戊酸酯(纯度98.5%)、泼尼卡酯(纯度99.2%)、哈西奈德(纯度99.2%)、阿氯米松双丙酸酯(纯度99.2%)、安西奈德(纯度99.7%)、氯倍他索丙酸酯(纯度99.0%)、氟替卡松丙酸酯(纯度99.2%)、莫米他松糠酸酯(纯度99.7%)、倍他米松双丙酸酯(纯度99.0%)、倍氯米松双丙酸酯(纯度99.8%)、氯倍他松丁酸酯(纯度98%)、醋酸甲地孕酮(纯度99.0%)、黄体酮(纯度99.2%)以上购自德国Dr. Ehrenstorfer公司、上海安谱实验科技股份有限公司、坛墨质检-标准物质中心。
乙腈(色谱纯) 德国Merck公司;甲酸(分析纯) 阿拉丁试剂(上海)有限公司;氯化钠、无水硫酸镁、中性氧化铝(均为分析纯) 广州化学试剂厂;PSA(40~60 µm,60A)、C18(40~60 µm)上海安谱科学仪器有限公司;超纯水由Milli-Q Element超纯水系统制备。
1.2 仪器与设备
Xevo TQ高效液相色谱-串联质谱联用仪 美国沃特世科技有限公司;JJ500Y电子天平 富阳腾辉电子科技有限公司;JP-C600超声波提取器 广州市吉普超声波电子设备有限公司;MS3涡旋混合器 德国艾卡公司;CM200-2氮吹仪 北京成萌伟业科技有限公司;3-18K高速台式冷冻离心机 德国Sigma公司;Milli-Q Element超纯水系统 美国Millipore公司。
1.3 方法
1.3.1 标准溶液的配制
标准储备液(1 mg/mL)配制:准确称取42 种类固醇激素标准物质各10.0 mg,用甲醇分别溶解定容至10.0 mL,于-18 ℃冷冻保存。
标准混合储备溶液(10 μg/mL)配制:分别取标准储备液1.0 mL混合,用甲醇定容至100 mL,于-18 ℃冷冻保存。
基质匹配混合标准溶液配制:临用时用40%乙腈溶液溶解的样品空白基质提取液稀释成0.5、1、5、10、25、50、100、250、500 μg/L系列质量浓度的标准混合工作溶液,用于制作标准曲线。
1.3.2 仪器条件
1.3.2.1 质谱条件
离子源:电喷雾离子源;扫描方式:正离子扫描;毛细管电压:2.83 kV;离子源温度:150 ℃;去溶剂温度:600 ℃;去溶剂气体流量:氮气,1 000 L/h;碰撞气:氩气;分段式多反应监测模式采集。42 种类固醇激素的保留时间、母离子、子离子以及对应的锥孔电压、碰撞能量见表1。
表1 42 种类固醇激素的质谱分析优化参数
Table 1 Optimized parameters for mass spectrometric analysis of 42 steroid hormones in the MRM mode
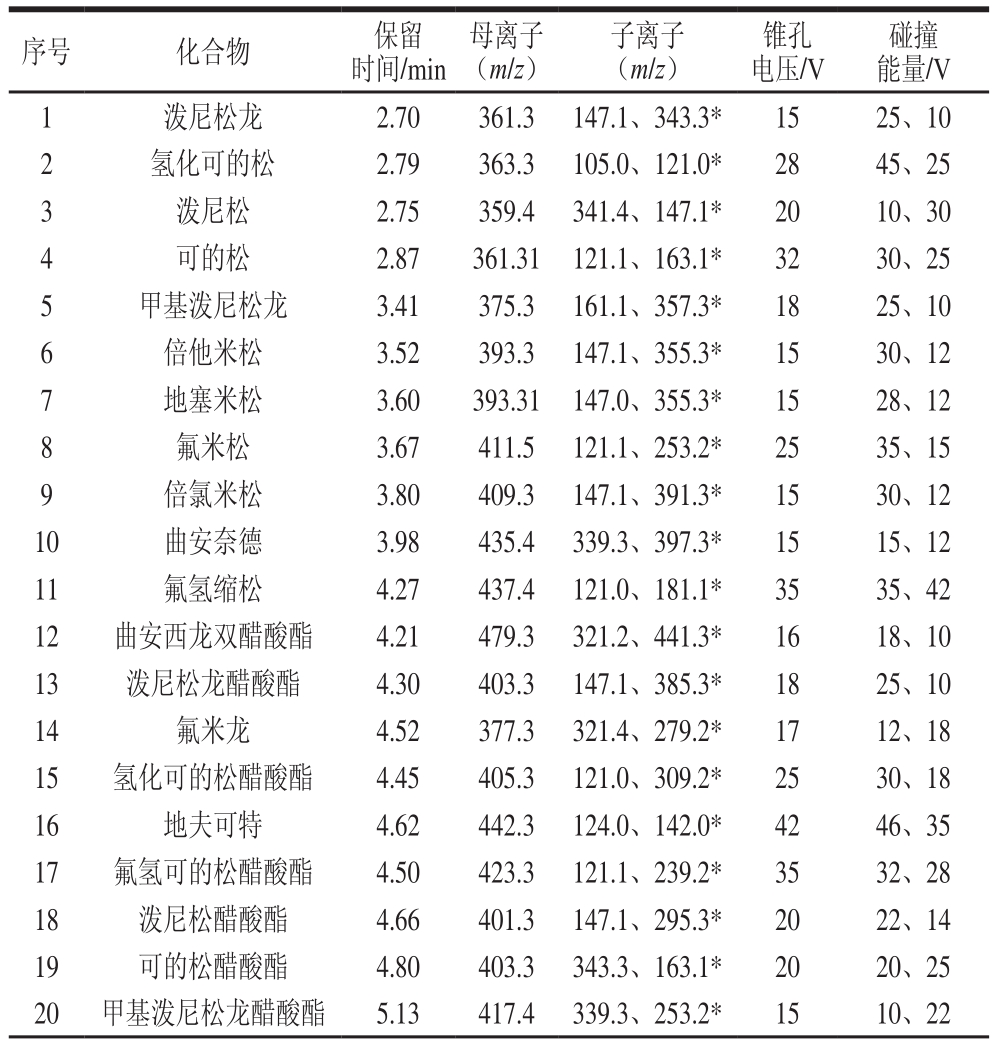
续表1

注:*.定量离子。
1.3.2.2 色谱条件
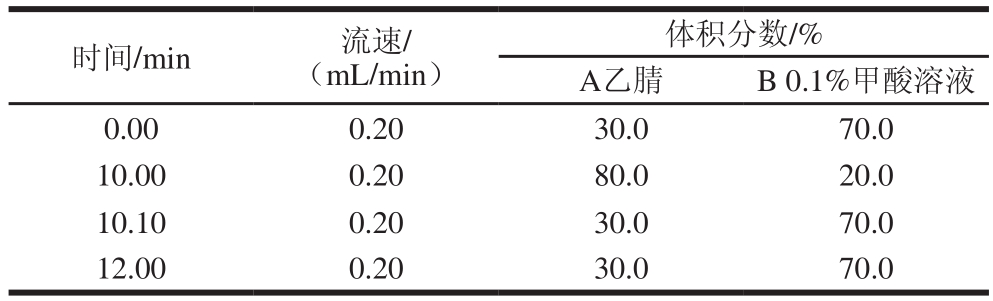
色谱柱:Waters ACQUITY UPLC®BEH C18(2.1 mm×100 mm,1.7 µm);进样量:10 μL;流动相:A为乙腈,B为0.1%甲酸溶液;柱温:30 ℃;梯度洗脱程序见表2。
表2 高效液相色谱梯度洗脱条件
Table 2 Gradient elution conditions of HPLC
1.3.3 样品前处理
1.3.3.1 奶粉
准确称取2 g(精确到0.01 g)试样于50 mL离心管中,加入4 mL 50 ℃温水溶解,涡旋混匀1 min,准确加入10 mL 0.1%甲酸-乙腈溶液,涡旋振荡2 min,超声提取15 min。加入1 g氯化钠和3 g无水硫酸镁,涡旋振荡2 min,9 000 r/min高速冷冻离心5 min。取上清液约8 mL置于装有50 mg C18、100 mg PSA、300 mg中性氧化铝和300 mg无水硫酸镁的另一离心管中,涡旋振动2 min,9 000 r/min高速冷冻离心3 min。准确移取5 mL上清液于10 mL离心管中,45 ℃氮气流吹干,用1 mL 40%乙腈溶液溶解残渣。经0.22 μm滤膜过滤,超高效液相色谱-串联质谱测定。
1.3.3.2 牛奶
准确称取5 g(精确到0.01 g)试样于50 mL离心管中,以下步骤同1.3.3.1节。
2 结果与分析
2.1 色谱条件的优化
选择一系列C18(ODS/SB/BEH)型色谱柱进行峰形比较,在相同色谱质谱条件下Waters ACQUITY UPLC®BEH C18(2.1 mm×100 mm,1.7 µm)柱更能够获得尖锐对称的优异峰形,选择性、灵敏度和稳定性好,确定其为本方法的色谱分离柱。Van Den Hauwe等[26]在分析糖皮质残留时,采用乙腈和不同体积分数的甲酸(0.05%~0.3%)溶液作为流动相,甲酸体积分数在0.05%~0.1%之间时,随着甲酸体积分数的增加,16 种糖皮质激素的响应值逐渐增加;但甲酸体积分数在0.1%~0.3%之间时,随着甲酸体积分数的增加,响应值又逐渐降低;本实验比较了乙腈-0.1%甲酸溶液、0.1%甲酸乙腈-0.1%甲酸溶液、乙腈-0.1%甲酸溶液(含5 mmol/L乙酸铵)的3 种流动相方案,测试100 ng/mL混合标准溶液在相同条件不同流动相体系中的响应值差异,结果以某种流动相体系与A流动相体系中42 种激素响应平均值的比值计算,绘制变化曲线(图1)。结果表明,流动相含有乙酸铵和体积分数过高的甲酸,反而会影响离子化效率,降低响应值,而乙腈-0.1%甲酸溶液流动相体系能提供最佳离子化条件,峰面积信号最强,灵敏度最高,符合以上结论。
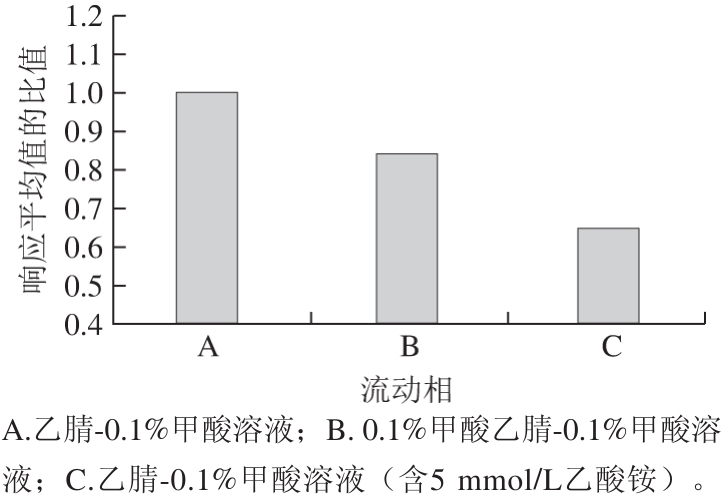
图1 不同流动相的比较
Fig. 1 Comparison of different mobile phases
2.2 质谱条件的确定
在电喷雾质谱、正离子监测模式下,分5 组将质量浓度为1 μg/mL的类固醇激素混合标准溶液以5 μL/min的流速直接注入质谱仪中进行一级质谱扫描,分别优化毛细管电压、锥孔电压、离子源温度、脱溶剂温度和流量获得各种化合物的母离子峰[M+H]+。根据欧盟2002/657/EC决议中有关质谱分析方法必须不少于4 个识别点的规定[27],选择各个待测化合物碰撞后所得丰度较高的两个子离子作为定量和定性离子,并确定其最佳碰撞能量的电压值,最终确定的母离子、子离子相关质谱参数见表1。由于同时测定的化合物较多,质谱在单位时间内获得的扫描次数较少,因此建立42 种目标化合物的分段式多反应监测模式质谱扫描方案,从而确保离子在准确定量的情况下仍有较高的灵敏度,并能有效减少化合物之间的相互干扰。42 种类固醇激素的分段式总离子流色谱图见图2。

图2 42 种类固醇激素的总离子流色谱图
Fig. 2 Total ion current (TIC) chromatograms of 42 steroid hormones
2.3 前处理条件的优化
2.3.1 提取溶剂的优化
大部分类固醇激素的极性较低,容易溶于脂肪含量较高的基质中,在弱极性或中等极性有机溶剂中有较高的溶解性,常用的提取溶剂包括乙醚、乙腈、乙酸乙酯、甲醇、叔丁基甲醚、正己烷、氯仿、二氯甲烷。赵文荣等[28]挑选了几种溶剂进行提取效果比对实验,结果显示正己烷和乙醚与水不分层,共萃物多,干扰严重;乙酸乙酯与水不互溶,程序复杂,容易导致基质损失;而乙腈较甲醇分层效果好,具有沉淀蛋白作用,响应高基质影响小。蛋白质和脂肪是奶粉和牛奶中的主要干扰物,高速冷冻离心能够分离沉淀部分脂肪,在提取过程中用适量的乙腈和含有甲酸的酸化乙腈[3]能够更好地去除杂质。因此本实验确定采用乙腈体系进行提取,并且比较了乙腈和0.1%甲酸-乙腈两种溶剂的提取效果(图3),结果表明,0.1%甲酸-乙腈作为提取溶剂的加标回收率稍高于乙腈,且提取效果优于后者。
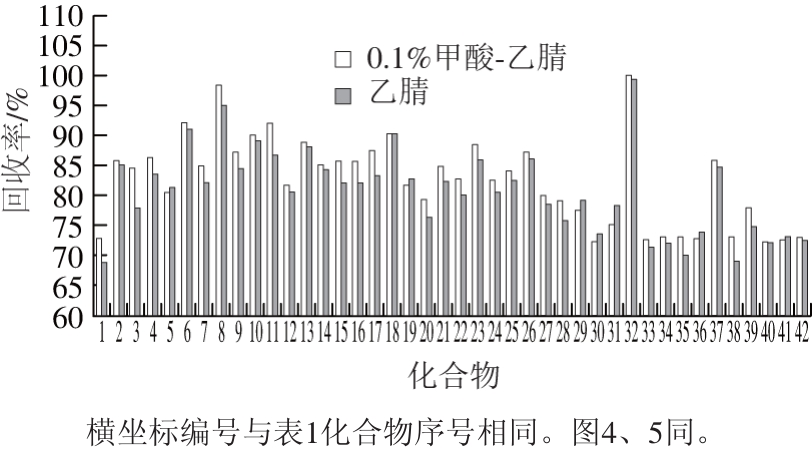
图3 不同溶剂对42 种类固醇激素的提取效果
Fig. 3 Extraction efficiencies of 42 steroid hormones with different solvents
2.3.2 净化填料的优化
PSA、C18是QuEChERS方法常用的吸附剂。PSA可有效去除乙腈提取液中的脂肪酸、糖类、酚类以及极性色素等成分,C18具有良好的除脂能力,并能吸附部分色素和维生素,但在实际操作中发现,对于脂肪含量较高的奶粉试样,提取液经C18和PSA净化后脂肪的去除效果并不理想。中性氧化铝对大部分目标物的吸附较少,因此可以作为净化剂进一步优化,净化后的溶液较之前清澈透亮,基线较平整,回收率在80%~120%之间,可以满足目标物痕量分析的要求。由于PSA、C18和中性氧化铝3 种净化填料,使用量少则达不到净化要求,使用量大则产生吸附,影响回收率,因此本实验考察各种填料的最佳使用量(图4~6),最终采用50 mg C18、100 mg PSA、300 mg中性氧化铝和300 mg无水硫酸镁的组合方案进行净化,结果显示,样品溶液澄清、基质干扰小,回收率佳。

图4 C18加入量对目标物回收率的影响
Fig. 4 Effects of C18dosage on the recovery of the target compounds
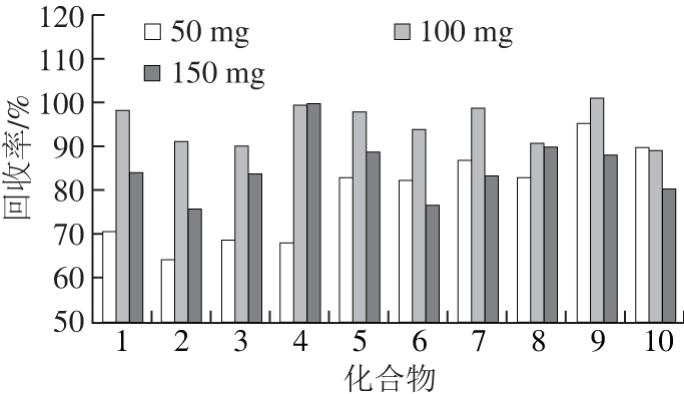
图5 PSA加入量对目标物回收率的影响
Fig. 5 Effects of PSA dosage on the recovery of the target compounds
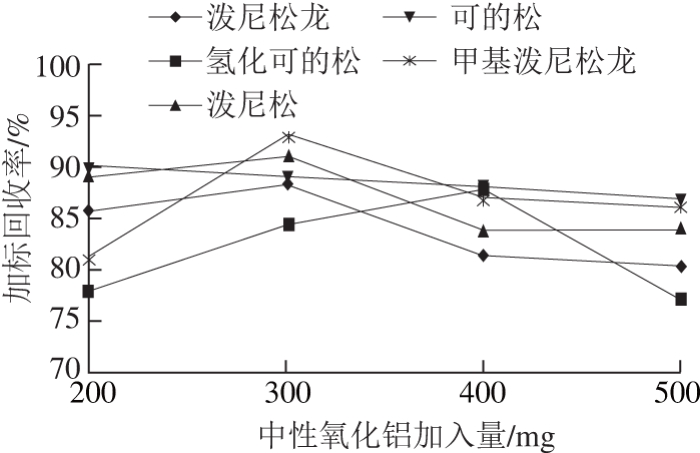
图6 中性氧化铝加入量对目标物回收率的影响
Fig. 6 Effects of alumina-N dosage on the recovery of the target compounds
2.3.3 定容液的优化
在多激素残留的同时定性定量分析中,定容液的极性与目标化合物的响应和峰形密切相关。本实验采用乙腈-水作为定容液,对两种溶液的比例进行考察。选择极性递增的4 个比例定容液,比较各种类型定容液的响应值及分离度差异。由图7可见,当定容液随着乙腈体积分数的升高,峰形逐渐变差,分离度下降。当乙腈体积分数为40%时,42 种激素的响应值最高,而且峰形优异,最终确定其为本方法的定容液。
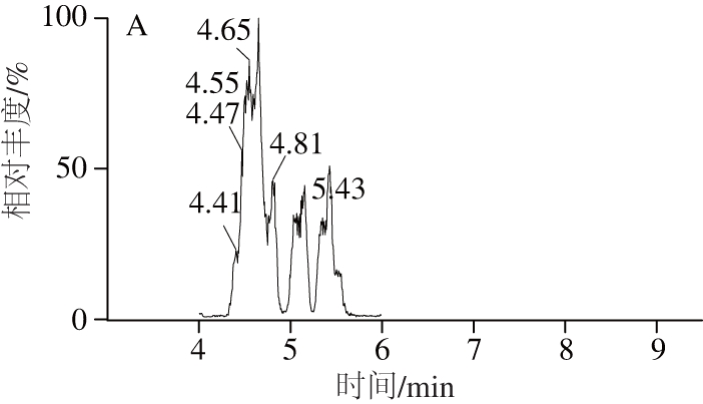

图7 不同定容液对峰形和响应值的影响
Fig. 7 Effects of different solvents on peak shape and response
2.3.4 基质效应的消除
基质效应是液相色谱-串联质谱测试中最常碰到的干扰效应,尤其对于基质复杂的样品(如动物源食品及生物样本)中表现得尤为明显,分为基质抑制效应和基质增强效应,是由基质成分和目标化合物在电喷雾离子源进行离子化时相互竞争导致的结果。基质效应可以通过比较待测物加入空白提取液中的响应值与待测物在纯溶剂中的响应值之比来确定其高低,该方法能够对基质效应进行定量计算和评价[29]。乳制品尽管经过乙腈沉淀蛋白、分散固相萃取后得到良好的净化效果,但仍然无法完全消除其基质效应。为保证最终结果的准确可靠,有必要引入基质匹配标准溶液的方法,尽可能降低基质效应造成的影响。
2.4 方法验证
2.4.1 专属性分析
取市售空白基质奶粉,该试样除了含有内源性黄体酮外,并未检出其他干扰性成分,作为空白样品,加标回收计算时应减去相应本底空白值。空白基质奶粉和42 种类固醇激素的空白样品添加(100 μg/kg)色谱图见图8。42 种类固醇激素分布在1~9 min之内,分离良好。
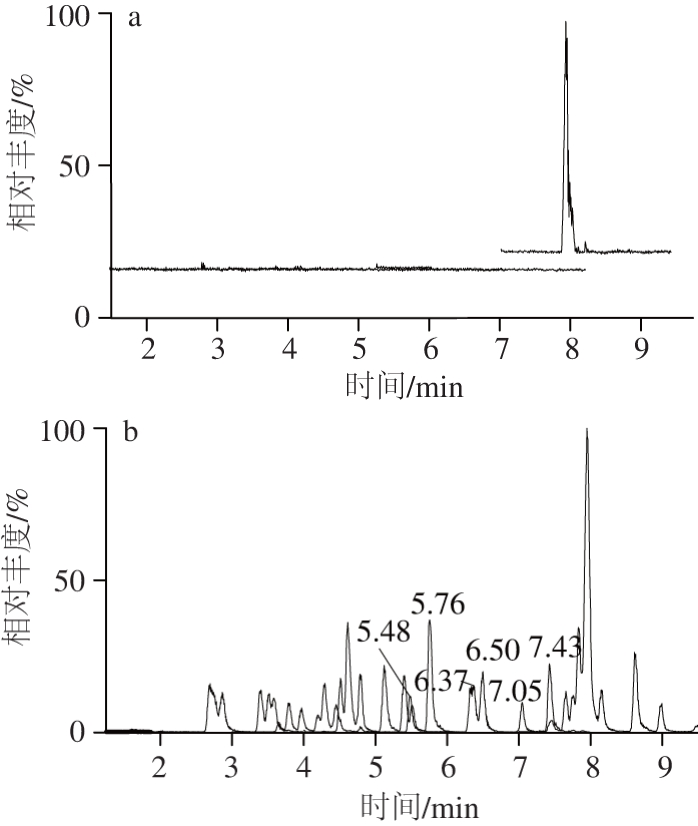
图8 空白奶粉(a)和空白奶粉添加(100 μg/kg)(b)色谱图
Fig. 8 Chromatograms of negative control (a) and milk powder spiked at 100 μg/kg (b)
2.4.2 线性和检出限
用空白样品基质提取液配制42 种类固醇激素的系列混合标准溶液,质量浓度分别为0、0.5、1、5、10、25、50、100、250、500 μg/L,在优化的实验条件下分离检测,以质量浓度(x)为横坐标,以定量离子的峰面积(y)为纵坐标制作标准曲线,得到目标化合物的线性回归方程和相关系数(r2)。如表3所示,42 种待测物的线性相关系数均大于0.99,表明各目标化合物在1~500 μg/L范围内呈良好的线性关系。
表3 42 种类固醇激素的线性范围、回归方程、相关系数、LOD及LOQ
Table 3 Linear ranges, regression equations, correlation coefficients,limits of detection (LODs) and limits of quantification (LOQs) of 42 steroid hormones
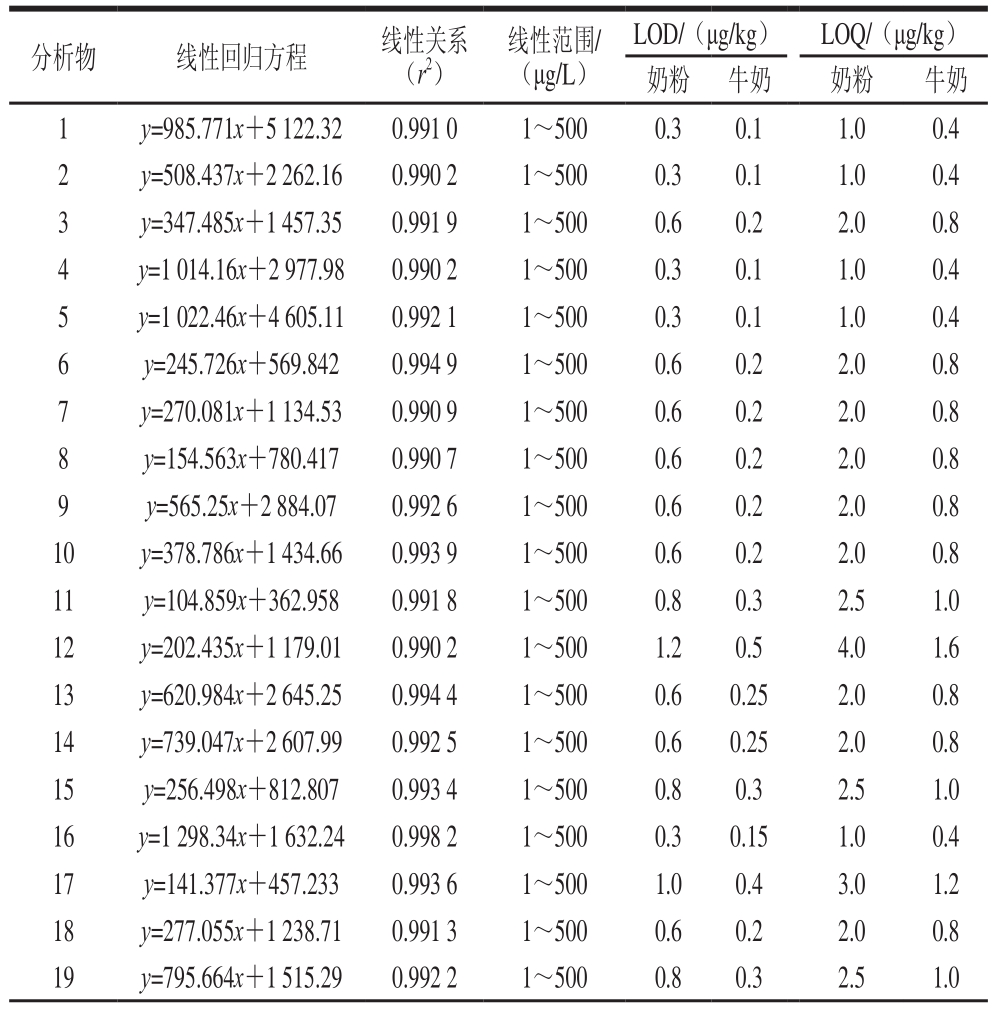
续表3
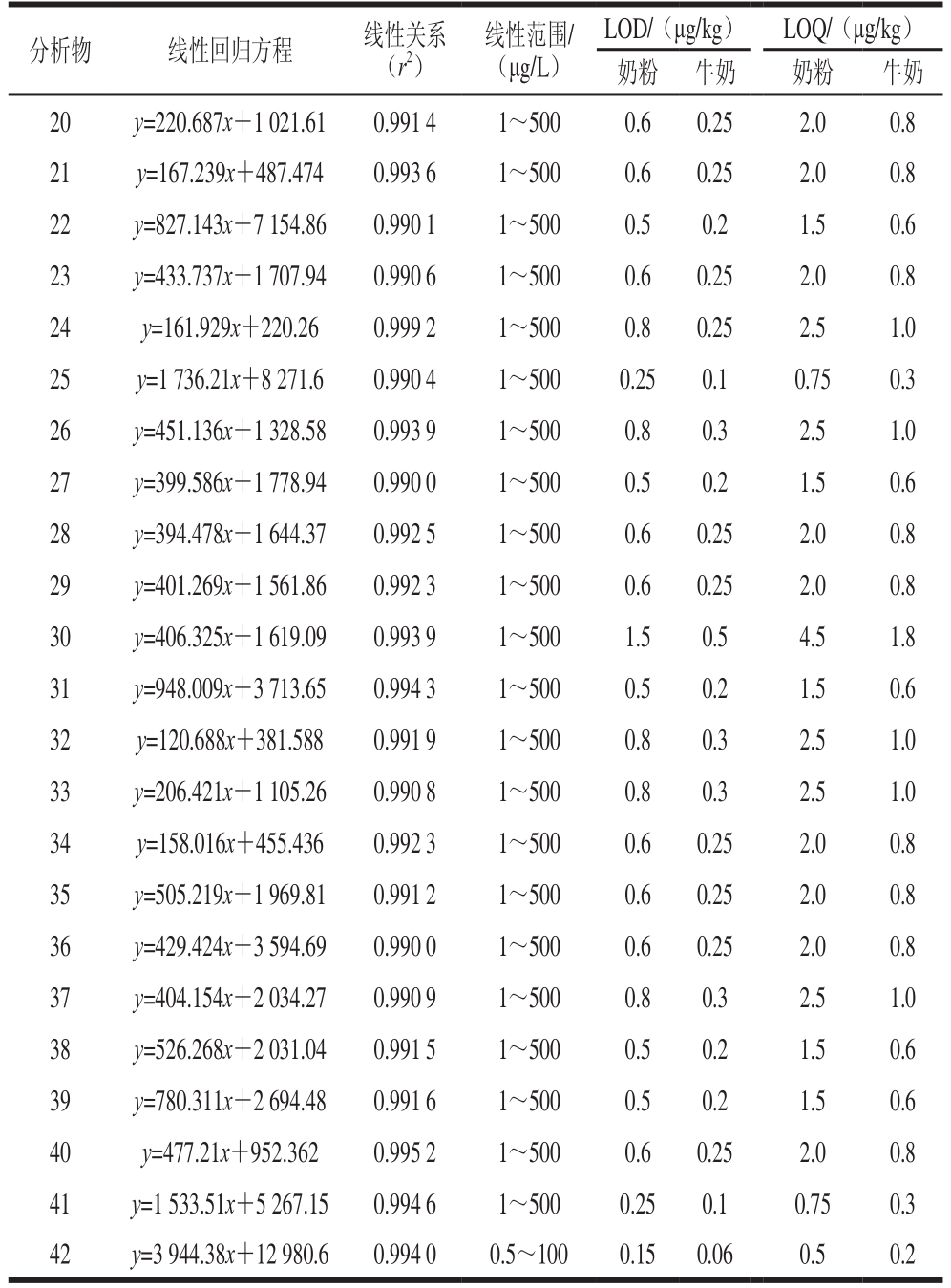
注:分析物序号对应表1化合物序号。
在空白奶粉和牛奶样品中添加一定质量浓度的混合标准溶液,将标准溶液与空白样品混合均匀后,按本研究建立的方法进行检测,以信噪比大于或等于10确定奶粉中各化合物的定量限(limit of quantitation,LOQ)为0.5~4.5 µg/kg,以信噪比大于或等于3确定各化合物的检出限(limits of quantification,LOD)为0.15~1.5 µg/kg;牛奶中各化合物的LOQ为0.2~1.8 µg/kg,LOD为0.06~0.5 µg/kg。
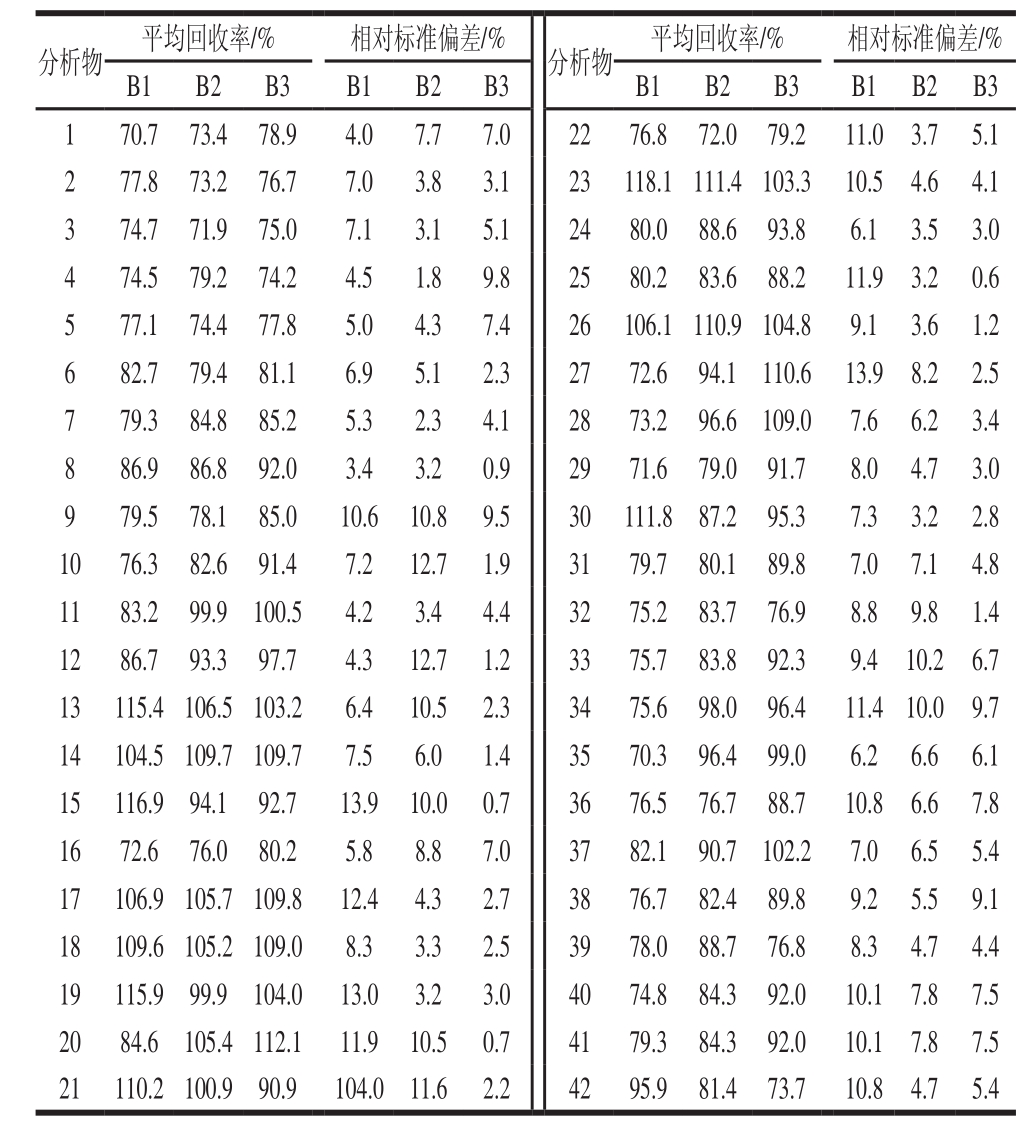
2.4.3 回收率和精密度实验结果
在奶粉和牛奶的空白样品中分别添加42 种类固醇激素混合标准溶液,奶粉的加标水平A1、A2、A3分别代表25、100、250 μg/kg,牛奶的加标水平B1、B2、B3分别代表10、40、100 μg/kg,每个水平平行测试6 次计算方法的平均回收率和精密度。如表4和表5所示,奶粉中42 种类固醇激素的加标回收率在70.7%~117.7%之间,相对标准偏差在0.7%~14.7%之间;牛奶的加标回收率在70.3%~118.1%之间,相对标准偏差在0.6%~13.9%之间。两种基质的平均加标回收率在70%~120%之间,相对标准偏差均不超过15%。结果表明本方法具有良好的准确度和精密度,符合分析要求。
表4 奶粉中42 种类固醇激素的平均回收率及精密度(n=6)
Table 4 Average recoveries and precisions (RSDs) of 42 steroid hormones in milk powder (n= 6)

注:分析物序号对应表1化合物。下同。
表5 牛奶中42 种类固醇激素的平均回收率及精密度(n= 6)
Table 5 Average recoveries and precisions (RSDs) of 42 steroid hormones in milk (n= 6)
2.5 实际样品的测定结果
分别对市场销售的24 个奶粉样品和24 个牛奶样品进行42 种类固醇激素残留量的检测,结果显示,48 个乳制品中除了黄体酮外均未检出其他41 种类固醇激素。奶粉中黄体酮的检出含量为0.6~23.8 μg/kg,检出率为95.8%;牛奶中黄体酮的检出含量为0.3~0.8 μg/kg,检出率为20.8%。黄体酮是卵巢黄体分泌的一种天然内源性孕激素,在体内对雌激素激发过的子宫内膜有显著形态学影响,为维持妊娠所必须[30],因此在动物源性的乳制品中具有一定比例的检出率。长期食用含有激素的食品可导致儿童性早熟,但关于黄体酮对婴幼儿健康产生的健康影响,目前尚未有确切的定论。
3 结 论
本实验采用QuEChERS前处理方法并结合超高效液相色谱-串联质谱技术,建立了乳制品中42 种类固醇激素残留量的检测方法。该方法前处理方法简单、简便有效、灵敏度高、选择性好、准确度和精密度高,回收率高且稳定,可用于实际样品的测定,为多激素残留的同时检测提供了新的检测途径。
参考文献:
[1] 徐锦忠, 张晓燕, 丁涛, 等. 高效液相色谱-串联质谱法同时检测鸡肉和鸡蛋中合成类固醇类激素和糖皮质激素[J]. 分析化学, 2009,37(3): 341-346. DOI:10.3321/j.issn:0253-3820.2009.03.006.
[2] 农业部. 动物性食品中兽药最高残留限量: 农业部公告第235号[R].2002-12-24.
[3] 崔晓亮, 邵兵, 赵榕, 等. 超高效液相色谱-串联电喷雾四极杆质谱法同时测定牛奶中12 种糖皮质激素的残留[J]. 色谱, 2006, 24(3): 213-217. DOI:10.3321/j.issn:1000-8713.2006.03.001.
[4] SILVA A C, EBRAHIMI-NAJAFADABI H, MCGINITIE T M, et al. Thermodynamic-based retention time predictions of endogenous steroids in comprehensive two-dimensional gas chromatography[J].Analytical and Bioanalytical Chemistry, 2015, 407(14): 4091-4099.DOI:10.1007/s00216-015-8627-0.
[5] RANDAZZO G M, TONOLI D, HAMBYE S, et al. Prediction of retention time in reversed-phase liquid chromatography as a tool for steroid identification[J]. Analytica Chimica Acta, 2016, 916: 8-16.DOI:10.1016/j.aca.2016.02.014.
[6] 万隽, 张晓光, 陈瑶. 糖皮质激素类药物对学龄前儿童CYP3A酶活性的影响[J]. 临床药学与研究, 2016, 27(11): 1473-1477.DOI:10.6039/j.issn.1001-0408.2016.11.11.
[7] LIAO K, MEI M, LI H, et al. Multiple monolithic fiber solidphase microextraction based on a polymeric ionic liquid with highperformance liquid chromatography for the determination of steroid sex hormones in water and urine[J]. Journal of Separation Science,2016, 39(3): 566-575. DOI:10.1002/jssc.201501156.
[8] HU Y, ZHANG M, TONG C, et al. Enrichment of steroid hormones in water with porous and hydrophobic polymer-based SPE followed by HPLC-UV determination[J]. Journal of Separation Science, 2013,36(20): 3321-3329. DOI:10.1002/jssc.201300663.
[9] CHA E, JEONG E S, CHA S, et al. Coupling of gas chromatography and electrospray ionization high resolution mass spectrometry for the analysis of anabolic steroids as trimethylsilyl derivatives in human urine[J]. Analytica Chimica Acta, 2017, 964: 123-133. DOI:10.1016/j.aca.2017.01.058.
[10] JANSSENS G, MANGELINCKX S, COURTHEYN D, et al.Simultaneous detection of androgen and estrogen abuse in breeding animals by gas chromatography-mass spectrometry/combustion/isotope ratio mass spectrometry (GC-MS/C/IRMS) evaluated against alternative methods[J]. Journal of Agricultural and Food Chemistry,2015, 63(34): 7574-7581. DOI:10.1021/acs.jafc.5b02746.
[11] CASALS G, MARCOS J, POZO O J, et al. Gas chromatographymass spectrometry profiling of steroids in urine of patients with acute intermittent porphyria[J]. Clinical Biochemistry, 2013, 46(9): 819-824.DOI:10.1016/j.clinbiochem.2013.03.001.
[12] JANSSENS G, MANGELINCKX S, COURTHEYN D, et al. The use of gas chromatography-mass spectrometry/combustion/isotope ratio mass spectrometry to demonstrate progesterone treatment in bovines[J]. Journal of Chromatography A, 2016, 1449: 129-140.DOI:10.1016/j.chroma.2016.04.074.
[13] 祝伟霞, 刘亚风. 液相色谱-串联质谱法快速测定婴幼儿配方奶粉中39 种激素残留[J]. 色谱, 2010, 28(11): 1031-1037. DOI:10.3724/SP.J.1123.2010.01031.
[14] FAHLBUSCH F B, HEUSSNER K, SCHMID M, et al. Measurement of amniotic fluid steroids of midgestation via LC-MS/MS[J]. Journal of Steroid Biochemistry & Molecular Biology, 2015, 152: 155-160.DOI:10.1016/j.jsbmb.2015.05.014.
[15] KEEVIL B G. LC-MS/MS analysis of steroids in the clinical laboratory[J]. Clinical Biochemistry, 2016, 49(13/14): 989-997.DOI:10.1016/j.clinbiochem.2016.04.009.
[16] LI C, WU Y L, YANG T, et al, Rapid simultaneous determination of dexamethasone and betamethasone in milk by liquid chromatography tandem mass spectrometry with isotope dilution[J]. Journal of Chromatography A, 2010, 1217(3): 411-414. DOI:10.1016/j.chroma.2009.12.015.
[17] HOLST B S, KUSHNIR M M, BERQUIST J. Liquid chromatographytandem mass spectrometry (LC-MS/MS) for analysis of endogenous steroids in the luteal phase and early pregnancy in dogs: a pilot study[J]. Veterinary Clinical Pathology, 2015, 44(4): 552-558.DOI:10.1111/vcp.12301.
[18] 黄丽英, 张晓波, 陈小珍, 等. UPLC-MS/MS法分析婴幼儿配方乳粉中12 种雌性激素[J]. 分析实验室, 2014, 33(1): 54-58. DOI:10.13595/j.cnki.issn1000-0720.2014.0012.
[19] 刘宏程, 李宁, 林涛, 等. 基质固相分散-超高效液相色谱-质谱检测器测定牛奶中9 种类固醇激素残留[J]. 色谱, 2015, 33(11): 1163-1168. DOI:10.3724/SP.J.1123.2015.06035.
[20] 钱叶飞, 贾昌平, 鲁辉, 等. UPLC-MS-MS快速检测中成药及保健食品中非法添加的38 种糖皮质激素[J]. 中国实验方剂学杂志, 2016,22(10): 60-66. DOI:10.13422/j.cnki.syfjx.2016100060.
[21] 李彦, 关丽丽, 牟妍, 等. QuEChERS-UPLC-MS/MS检测禽畜组织中金刚烷胺残留[J]. 检测分析, 2013, 24(23): 37-40. DOI:10.3969/j.issn.1005-6521.2013.23.010.
[22] CALATAYUD-VERNICH P, CALATAYUD F, SIMO E, et al.Efficiency of QuEChERS approach for determining 52 pesticide residues in honey and honey bees[J]. MethodsX, 2016, 3: 452-458.DOI:10.1016/j.mex.2016.05.005.
[23] PAYA P, ANASTASSIADES M, MACK D, et al. Analysis of pesticide residues using the quick easy cheap effective rugged and safe (QuEChERS) pesticide multiresidue method in combination with gas and liquid chromatography and tandem mass spectrometric detection[J]. Analytical and Bioanalytical Chemistry, 2007, 389: 1697-1714. DOI:10.1007/s00216-007-1610-7.
[24] BRUZZONITI M C, CHECCHINI L, DE CARLO R M, et al.QuEChERS sample preparation for the determination of pesticides and other organic residues in environmental matrices: a critical review[J].Analytical and Bioanalytical Chemistry, 2014, 406(17): 4089-4116.DOI:10.1007/s00216-014-7798-4.
[25] LOZOWICKA B, RUTKOWSKA E, JANKOWSKA M. Influence of QuEChERS modifications on recovery and matrix effect during the multi-residue pesticide analysis in soil by GC/MS/MS and GC/ECD/NPD[J]. Environmental Science and Pollution Research, 2017, 24(8):7124-7138. DOI:10.1007/s11356-016-8334-1.
[26] VAN DEN HAUWE O, DUMOULIN F, ELLIOTT C, et al.Detection of synthetic glucocorticoid residues in cattle tissue and hair samples after a single dose administration using LC-MS/MS[J].Journal of Chromatography B, 2005, 817(2): 215-223. DOI:10.1016/j.jchromb.2004.12.006.
[27] The Commission of the European Communities 2002/657/EC.Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC Concerning the performance of analytical methods and the interpretation of results[S]. Official Journal of the European Communities, 2002, L221: 8-36.
[28] 赵文荣, 李刚, 李宁, 等. 高效液相色谱-串联质谱法快速检测牛奶中13 种糖皮质激素的残留量[J]. 农产品加工: 学刊, 2013(5): 66-69.DOI:10.3969/jissn.1671-9646(X).2013.05.047.
[29] MATUSZEWSKI B K. Standard line slopes as a measure of a relative matrix effect in quantitative HPLC-MS bioanalysis[J].Journal of Chromatography B, 2005, 830(2): 293-300. DOI:10.1016/j.jchromb.2005.11.009.
[30] 李佩斯. 国内外婴幼儿配方奶粉激素含量比较[J]. 中国乳业, 2014,52(3): 52-54. DOI:10.16172/j.cnki.114768.2014.03.001.