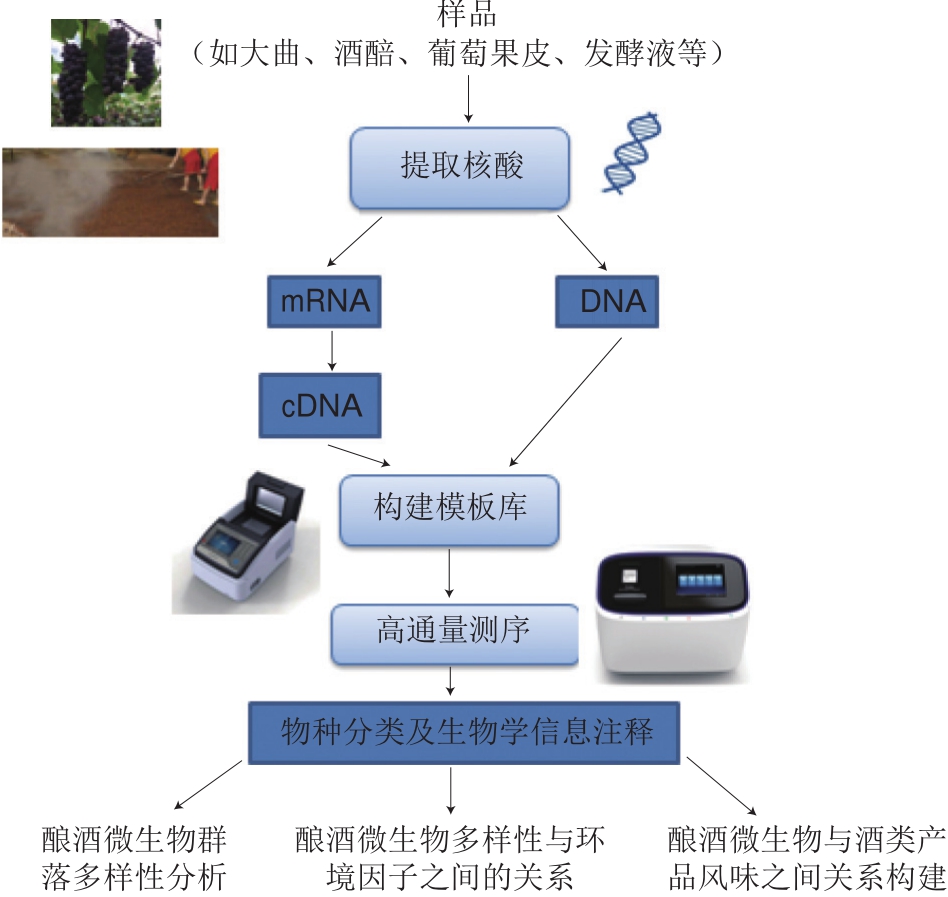
图1 高通量测序技术流程图(针对酿酒微生物多样性研究)[10,12-15]
Fig.1 Flow chart of high-throughput sequencing technology applied to analyze the diversity of microbial communities involved in the fermentation of alcoholic beverages[10,12-15]
中国酒类产品种类丰富,包括白酒、米酒、黄酒、啤酒及葡萄酒等,均具有悠久的酿造历史,各具独特的风味。其中,茅台酒、绍兴黄酒及烟台张裕葡萄酒等产品更是以其独特的风味闻名于世。据报道,2009年,中国白酒年消费量已超过40亿 t[1],深受国内外消费者的喜爱。中国酒类产品的独特风味与其酿造过程中微生物的消长演替相关,细菌、酵母菌、霉菌等微生物在其生产及风味形成过程中发挥了至关重要的作用。酿酒原料、酿造工艺、地理位置及气候条件等因素会影响酿酒微生物群落结构,因此,酿酒微生物多样性与中国各酒类产品的风味特征息息相关[2]。
自1904年日本学者齐藤研究中国绍兴酒中酿酒微生物的多样性至今已有100多年的历史[3]。起初对酿酒微生物多样性的研究主要依赖于传统的技术手段,得到的研究结果具有一定的局限性。现代分子生物技术的发展促进了越来越多不依赖于培养的技术的发展与应用。对葡萄酒酿造过程中微生物多样性的相关研究发现,原位杂交技术[4]、实时定量聚合酶链式反应(real-time quantitative polymerase chain reaction,qPCR)[5]、基因芯片技术[6]、变性凝胶梯度电泳技术[7-8]及高通量测序技术[9]等不依赖于培养的技术,的确有助于对处于存活但不可培养状态的菌群或细胞浓度较低菌群的检测、鉴定及定量分析[10-11]。近年来,由于高通量测序技术具有测序时间短、测序通量高的优势,国内外研究者将该技术应用于酿酒微生物多样性的研究中,促进了研究者对于酿酒微生物多样性的认识。本文在介绍高通量测序技术的基础上,分析其在酿酒微生物多样性研究中的应用情况,旨在为中国酒类产品的发酵机理、质量与风味控制等相关领域的深入研究提供理论指导。
高通量测序又称下一代测序、大规模测序等,由第一代测序技术即Maxam-Gilbert和Sanger测序发展而来[12]。与第一代测序技术相比,高通量测序技术可以在较短的时间内对数以千计甚至百万计的序列进行检测分析。因此,高通量测序技术不仅有利于复杂微生物群落多样性的快速分析,而且可以检测到低存在率的微生物以及存活但不可培养的微生物,从而提供更为全面的微生物群落信息。
近年来,结合本课题组采用高通量测序技术对酒样(如白酒酿造过程中的大曲、葡萄酒酿造过程中的发酵液等)中微生物群落多样性进行的研究可知,高通量测序技术流程一般包括采样、提取及纯化核酸序列、构建文库、测序、原始序列信息比对注释、数据分析等步骤(图1)[10,12-15]。整个技术流程中,关于高通量测序平台的选择和数据分析方法的使用两方面的报道较少,而其对微生物多样性的研究十分重要,因此本文在技术部分将着重介绍这两方面的内容。
图1 高通量测序技术流程图(针对酿酒微生物多样性研究)[10,12-15]
Fig.1 Flow chart of high-throughput sequencing technology applied to analyze the diversity of microbial communities involved in the fermentation of alcoholic beverages[10,12-15]
目前可采用的高通量测序平台主要包括Roche 454焦磷酸测序平台、Illumina测序平台、SOLiD测序平台、Ion Torrent测序平台和PacBio SMRT测序平台(第三代测序平台)。这五大主要测序平台在测序原理、测序试剂、序列读取长度、测序数量等方面都存在差异(表1)[9,16-20]。由于酿酒微生物特定核酸片段序列长度一般在1 000 bp以内,因此在微生物多样性研究中较常采用的为Roche 454和Illumina测序平台。
高通量测序平台输出的是原始序列数据reads,研究者一般将其分类鉴定为可操控分类单元(operational taxonomic units,OTUs)后再进行统计分析。OTUs的获取主要通过与已建立的基因数据库比对以获得分类学注释。但是,数据库中未储备信息的菌种无法得到分类鉴定。目前,常用的基因数据库有GenBank[21]、RDP[22]、SILVA[15]、Greengenes[23]和UNITE[24]等。近年来研究者广泛使用Mothur[25]和QIIME[26]进行分类学注释及后续统计分析。在后续的分析中,常用的多元统计学方法有聚类分析、判别分析、排序分析、相关性分析和差异性分析等[27]。采取合适的数据分析策略可以准确反映酿酒微生物群落的多态性,溯源酒类产品的风味物质,把握酿酒微生物与环境因子的相互作用以及调控其中的健康因子和有害物种等[28]。在酿酒微生物多样性分析研究中常用的数据分析方法包括主成分分析(principal component analysis,PCA)、典型相关分析(canonical correlation analysis,CCA)及相对丰度热图。
表1 5 种高通量测序平台的测序原理、读取序列长度和优缺点[9,16-20]
Table1 Principle, sequencing length, merits and drawbacks of fi ve different high-throughput sequencing platforms[9,16-20]

注:各测序平台对应的测序模板结构图参考华大基因(http://www.genomics.cn/index)及文献[18],分别为含有DNA模板的微型有孔小珠(Roche 454)、桥式结构(Illumina)、Ion测序芯片(Ion Torrent)、ZMW模板(PacBio SMRT)、含DNA模板的P1磁性小珠(SOLiD)。
测序平台名称平台类别测序模板测序原理检测方法典型序列读取长度测序通量大小及测序时间测序错误率/% 优点 缺点Roche 454 第二代测序 焦磷酸测序,qPCR,边合成DNA分子边测序强度≤1 000 bp35~700 Mb、10~23 h0.4~1.5通量高,读取序列长度较长荧光测序价格昂贵Illumina 第二代测序
焦磷酸测序,qPCR,边合成DNA分子边测序强度≤1 000 bp35~700 Mb、10~23 h0.4~1.5通量高,读取序列长度较长荧光测序价格昂贵Illumina 第二代测序 合成测序,桥式PCR,边合成DNA分子边测序图像采集荧光标记150~300、300~600 bp最大可输出600 Gb碱基信息、运行时间最长可达11 d 0.5~2.0 通量高,测序价格较低读取序列长度较短Ion Torrent 第二代测序
合成测序,桥式PCR,边合成DNA分子边测序图像采集荧光标记150~300、300~600 bp最大可输出600 Gb碱基信息、运行时间最长可达11 d 0.5~2.0 通量高,测序价格较低读取序列长度较短Ion Torrent 第二代测序 合成测序,qPCR,边合成DNA分子边测序氢离子强度 400 bp 1 Gb、2 h 1~3读取长度中等,利于对小型基因组的测序研究精确度低PacBio SMRT 第三代测序
合成测序,qPCR,边合成DNA分子边测序氢离子强度 400 bp 1 Gb、2 h 1~3读取长度中等,利于对小型基因组的测序研究精确度低PacBio SMRT 第三代测序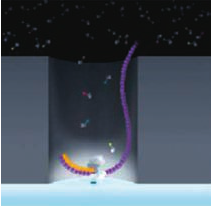 单分子实时测序,边合成DNA分子边测序荧光标记 7 kb最大可输出90 Mb碱基信息、1 h 13~15读取序列长度长,无需PCR扩增,精确度较高通量低SOLiD 第二代测序
单分子实时测序,边合成DNA分子边测序荧光标记 7 kb最大可输出90 Mb碱基信息、1 h 13~15读取序列长度长,无需PCR扩增,精确度较高通量低SOLiD 第二代测序 DNA连接酶,连接测序 荧光标记 75 bp最大可输出200 Gb碱基信息、运行时间最长可达7 d 0.01 精确度最高 读取序列长度短
DNA连接酶,连接测序 荧光标记 75 bp最大可输出200 Gb碱基信息、运行时间最长可达7 d 0.01 精确度最高 读取序列长度短
1.2.1 PCA
PCA是多元统计学分析方法的一种。主要是通过对大量的样本原始数据进行降维处理,从而分析样本之间差异性,并且可以对不同样本进行归类分析。PCA可使用SPSS、CANOCO等软件进行分析,其分析结果一般可通过得分图和载荷图体现。前者可以反映出各样本之间的差异性,而后者则可以反映出导致样本间产生差异性的具体原因[29-32]。PCA将测序数据反映在坐标轴行与行、行与列及列与列之间,直接反映其获得的主成分得分及变量得分,可为其他分析提供基础。但是,PCA中各样本间关系仅通过散点间距衡量,结果判断带有较强的主观性。本课题组采用高通量测序方法对3 种酱香型白酒中细菌微生物群落结构进行研究,采用PCA可以区分出3 种酱香型白酒大曲,并指出了产生差异的原因[33]。
1.2.2 CCA
CCA是利用综合变量之间的相关关系来反映两组指标间整体相关性的多元统计分析方法。其基本原理是:基于从总体上需要把握两组指标之间的相关关系,分别从两组变量中提取有代表性的两个综合变量U1和V1(分别为两个变量组中各变量的线性组合),利用这两个综合变量之间的相关关系来反映两组指标之间的整体相关性。CCA可以通过CANOCO软件对数据进行处理分析。借由CCA能够反映出酿酒微生物与环境因子之间的响应关系[34-35]。该方法不仅可以通过双标图反映物种与样本、样本与环境、物种与环境之间的关系,而且还可以直接通过三标图反映这三者之间的关系,降低干扰因素产生的方差。但是,CCA仅表明了其三者之间的一种趋势关系,结果判定主观性较强。Zheng Xiaowei等[34]运用CCA阐述了汾酒大曲中微生物多样性与环境因子(酸度、水分含量及温度等)之间的关系,其分析结果有助于控制汾酒酿造过程。
1.2.3 相对丰度热图
相对丰度热图可以反映样品之间的相似性以及群落结构的相似性[36]。在相对丰度热图中,每一个色块代表一个样品的物种分类学信息(相对丰度)。一般而言,样品横向排列,物种分类学信息纵向排列。相对丰度热图可以采用R软件(http://www.r-projict.org/)或HemI软件(http://hemi.biocuckoo.org/down.php)进行绘制。除此之外,不同测序公司也会基于高通量测序所得的原始序列数据,为研究者免费提供某些数据分析服务,其中也包括相对丰度热图。相对丰度热图不仅能够对物种进行聚类分析,并且注释有详细的物种丰度信息,研究者借此即可直观地对样本中的物种进行分析。Tang Jing等[36]采用相对丰度热图反映出了两种酱香型白酒大曲中细菌群落多样性的异同,并在此基础上进一步分析了其与酱香型白酒风味物质间的关系。
近年来,高通量测序技术在酒类产品酿酒微生物多样性的研究中得到了越来越多的关注和应用,涵盖了白酒、葡萄酒和其他酒类(表2)。其应用主要集中在对酿酒微生物进行分类鉴定、监测发酵过程中酿酒微生物群落动态变化、追溯酒类风味物质以及辨别酒类产品是否掺假等方面[2]。
在白酒领域,高通量测序技术多用于研究酒曲中酿酒微生物的多样性,对于窖泥和酒醅也有相关报道。高通量测序技术在白酒领域主要集中于对细菌多样性的分析,酒样主要为酱香、清香、浓香3 种香型。本课题组主要针对酱香型白酒大曲中微生物多样性进行研究,采用Roche 454焦磷酸测序平台对3 种酱香型白酒大曲中细菌菌群进行了测序,发现在3 种酱香型大曲中共检测到35 种细菌科,并且首次在酱香型大曲中报道了芽孢乳杆菌科、链霉菌科、假单胞菌科、多孢放线菌科及柄杆菌科等细菌。同时,通过采用PCA作图,进一步分析出3 种酱香型大曲差异性的原因[33]。运用高通量测序技术对酱香型白酒不同发酵轮次酒中微生物多样性的研究也有报道[37-38]。本课题组采用高通量测序技术比较了不同轮次酒醅中的细菌与真菌群落结构的异同:发现酱香型白酒发酵过程3 轮次酒醅中细菌优势菌属为乳杆菌属和芽孢杆菌属,真菌优势菌属为嗜热真菌属和嗜热子囊菌属;而发酵7 轮次酒醅中细菌优势菌属为盐单胞菌属,真菌优势菌属为隐球菌属[37]。此外,Zhang Xiuhong等[13]采用Roche 454 GS FLX测序平台,对清香型白酒不同大曲中细菌群落结构进行了研究,结果表明清香型白酒不同大曲中细菌群落结构有较大差异。Zheng Qi等[39]采用Illumina测序平台探究了浓香型白酒不同年份窖池中窖泥微生物群落多样性及其与风味成分间的关系。
在葡萄酒领域,高通量测序多用于研究发酵过程中微生物的多样性及动态变化,对葡萄表面的微生物多样性也有相关报道。Portillo等[40]采用Ion Torrent PGM测序平台对西班牙Priorat地区不同葡萄园中歌海娜和佳丽酿葡萄表皮上细菌群落多样性进行了研究,并研究其与地理环境间的关系。结果表明厚壁菌门(Firmicutes)、变形杆菌门(Proteobacteria)和放线菌门(Actinobacteria)为主要的优势菌,且地理环境分布对发酵过程中的细菌群落结构影响明显。Wang Chunxiao等[10]采用传统的分离鉴定技术、高通量测序技术、变性梯度凝胶电泳技术和qPCR技术等不依赖于培养的技术,对产自西班牙Priorat地区3 个葡萄园中的歌海娜和佳丽酿葡萄发酵过程中的酵母菌群落结构进行了相关研究。结果表明,与其他方法相比,高通量测序技术虽无法进行定量检测,但可检测到绝大多数的非酿酒酵母菌属。
高通量测序技术还被应用于黄酒、米酒、安第斯吉开酒(chicha)等其余酒类领域中,已成功揭示了其各自发酵液或酒曲中的微生物多样性。Xie Guangfa等[41]采用Illumina HiSeq 2000测序平台从门、属分类学水平对不同发酵时间段绍兴黄酒酿酒微生物多样性进行了研究,结果表明发酵初期和后期其微生物群落结构截然不同,优势微生物的种类和数量发生明显变化。Mendoza等[21]采用Roche 454 GS Junior测序平台从安第斯吉开酒发酵过程中检测出超过100 种酵母菌,其中超过一半的酵母菌归属于丝状真菌属。
可见,虽然高通量测序技术被应用于不同的研究领域和样本,但目前针对酿酒微生物多样性分析相关研究所使用的测序目标片段、方法和平台具有相似性。而且,对于相同的酿酒微生物,采取不同的测序目标片段或方法都会影响测序结果的准确度及精确度[10,42-44]。一般对于原核微生物常选用的测序片段为16S rDNA V1~V9各变异区片段,而对于真核微生物而言,常选取的测序片段为18S rDNA、26S rDNA D1/D2区和内部转录间隔区(internal transcribed spacer,ITS)等。数据库的选用也会影响微生物鉴定的准确性,本课题组利用RDP、SILVA和GenBank数据库对葡萄酒中的真核微生物鉴定分析的对比结果表明,GenBank数据库更适用于真核微生物尤其是酵母菌的鉴定:其可以实现种水平上的鉴定;并且具备较高的菌种鉴定置信度,利于准确的种水平上菌株的鉴定[10]。目前,原核微生物的相关研究中主要选用的数据库为RDP和SILVA,但是尚鲜有相关报道对这两个数据库进行比对,本课题组拟在后期工作中开展相关研究。
聚焦至不同酒类的发酵过程,高通量测序的结果表明了更明显的微生物组成差异。目前,采用高通量测序技术对酱香型白酒微生物多样性的研究主要集中于揭示其细菌群落结构方面,测序发现主要的微生物为芽孢杆菌(Bacillaceae)和乳酸菌属(Lactobacillus)等。而对酱香型白酒中真核微生物群落结构也有相关报道,发现了嗜热真菌属(Thermomyces)、热子囊菌属(Thermascus)、隐球酵母属(Cryptococcus)、青霉菌属(Penicillium)等[14,33,36-38]。在浓香型白酒发酵过程中,发现厚壁菌属(Firmicutes)、放线菌属(Actinobacteria)、醋酸杆菌属(Acetobacter)、乳酸菌属(Lactobacillus)、甲烷杆菌属(Methanobacterium)等为主要的微生物[12,39,45-47]。在清香型白酒发酵过程中报道的主要微生物为厚壁菌属(Firmicutes)和放线菌属(Actinabacteria)[13,48]。不同白酒中微生物群落结构的差异是赋予白酒不同香型特征的重要原因。葡萄酒本土风味的形成与其在发酵过程中相关酵母菌属的消长演替密切相关。研究发现葡萄酒发酵过程中主要的酵母菌属包括酿酒酵母属(Saccharomyces)、有孢汉逊酵母属(Hansenispora)、汉生酵母属(Hansenula)、毕赤酵母属(Pichia)、假丝酵母属(Candida)等。而乳杆菌属(Lactobacillus)是葡萄酒发酵过程中主要的细菌微生物[10,49-53]。米酒作为中国传统的发酵食品之一,通过对其微生物多样性的研究发现,米酒中主要的微生物为芽孢杆菌属(Bacillaceae)、乳杆菌属(Lactobacillus)、乳球菌属(Lactococcus)、高温放线菌属(Thermoactinomyces)等[54-55]。高通量测序技术揭示,不同酒类的微生物组成差异主要表现在两个方面:1)主要微生物菌属(种)的种类和比例差异;2)小类微生物菌属(种)及未知菌属(种)的种类和比例差异。如具有复杂工艺的酱香型白酒酿造过程中发现了很多小类微生物菌属和未知菌属,虽然它们在微生物群落结构中所占比例较小,通过其他技术未必能分离出来,但其构成了酱香型白酒独特微生物组成特征不可忽视的一面。微生物的组成差异受到多种因素的影响,首先是酒类工艺过程,其次是同种酒类工艺在不同地区的酿造过程中发现的微生物组成差异,体现了微生物分布的地域性。
表2 高通量测序技术在酿酒微生物多样性研究中的应用情况
Table2 Recent applications of high-throughout sequencing technology to analyze the diversity of microbial communities involved in the fermentation of alcoholic beverages

样本 研究目标 测序片段(片段长度/bp)测序平台 采用的数据库 分析软件(分析方法)发表时间白酒领域酱香型白酒酒醅酱香型白酒中细菌群落多样性,不同轮次酒与环境因子(如温度)间的关系细菌:16S rDNA V4变异区(588~671)真菌:ITS区(500~750 )Roche 454 Greengenes QIIME(Chao1指数、Shannon指数等)2017[37]酱香型白酒酒醅酱香型白酒大曲酱香型白酒大曲酱香型白酒堆积及窖池发酵微生物群落多样性,物种与样品间的关系酱香型白酒大曲中细菌群落多样性及其在制作大曲、堆积发酵、窖池发酵阶段动态变化过程3 种酱香型白酒大曲中细菌群落多样性的异同细菌:16S rDNA(1 540)真菌:ITS区(500~750)Illumina MiSeq GenBank 上海美吉生物医药科技有限公司提供 2017[38]细菌:16S rDNA V6变异区(829~857)Illumina GAll RDP R软件(Shannon指数、Simpson指数、相对丰度热图、PCA、维恩图等)2015[14]细菌:16S rDNA V3~V5变异区(436~754)Roche 454 GS FLX+ Greengenes QIIME、Mothur(Shannon指数、Simpson指数、相对丰度热图、PCA等)2017[33]酱香型白酒大曲两种酱香型白酒大曲中细菌微生物群落的异同 细菌:16S rDNA V4变异区(588~671)Illumina MiSeq RDP QIIME、UPARSE、R软件(Chao1指数、Shannon指数、相对丰度热图)2017[36]清香型白酒大曲3 种清香型白酒大曲中细菌群落组成细菌:16S rDNA V1~V3变异区(61~106)Roche 454 GS FLX SILVA Mothur、Chopseq、Plot-faction(Chao1指数、Shannon指数、相对丰度热图等)2014[13]清香型白酒大曲清香型白酒大曲中微生物优势菌株变化细菌:16S rDNA V3~V4变异区(436~671)真菌:ITS1区(350)Illumina MiSeq GenBank QIIME(OTU比例图等)2016[48]浓香型白酒酒醅酿造季节对浓香型白酒酒醅中细菌群落多样性的影响 细菌:16S rDNA V4变异区(588~671)Illumina MiSeq RDP QIIME、USEARCH、Mothur、R软件(Chao1指数、ACE指数、Shannon指数、Simpson指数、相对丰度热图、聚类分析等)2016[12]浓香型白酒大曲、窖泥及发酵谷物浓香型白酒发酵过程中大曲、窖泥及谷物中原核微生物群落多样性细菌:16S rDNA V4变异区(588~671)Illumina MiSeq RDP QIIME、SPSS、R软件(Chao1指数、Shannon指数、PCoA、相对丰度热图等)2017[45]浓香型白酒窖泥浓香型白酒不同年份窖池中窖泥微生物群落多样性及其与风味成分间的关系细菌:16S rDNA V4变异区(588~671)Illumina MiSeq 基于QIIME软件的核糖体核苷酸数据库QIIME、UPARSE(相对丰度热图、系统进化树等)2015[39]浓香型白酒窖泥浓香型白酒窖泥微生物群落多样性及其与窖池环境之间的关系 细菌:16S rDNA V4变异区(588~671)Illumina MiSeq RDP QIIME、SPSS、CANOCO(Chao1指数、PCoA、CCA、相对丰度热图等)2016[46]浓香型白酒大曲16S rDNA克隆文库法与高通量测序技术分析浓香型白酒大曲中细菌微生物群落的异同细菌:16S rDNA(1 540)Illumina MiSeq RDP BiodaP、Ucluct(Shannon指数、相对丰度热图等)2015[47]葡萄酒领域葡萄醪产自西班牙Priorat地区不同葡萄园的歌海娜和佳丽酿酿酒葡萄表皮细菌群落多样性细菌:16S rDNA V4变异区(588~671)Ion Torrent PGM RDPU、ChimeraSlayer QIIME、PRIMER v6、FastTree(Chao1指数、Shannon指数、Simpson指数、相对丰度热图、维恩图等)2016[40]葡萄发酵液西班牙不同葡萄园中的歌海娜和佳丽酿酿酒葡萄酵母群落多样性酵母:26S rDNA D1/D2区(600)Roche 454 FLX GenBank Estimate S v9.1.0(相对丰度热图、Jaccard聚类分析等)2015[10]葡萄发酵液葡萄牙不同地区葡萄不同发酵阶段中微生物群落的变化情况细菌:16S DNA V6变异区(829~857)真菌:ITS2区(400);26S rDNA D2区(240)Roche 454 RDP、GenBank、SILVA SPSS(Chao1指数)、SIMPER(PCoA、相对丰度热图)2015[44]葡萄发酵液意大利两种酿酒葡萄自然发酵过程中真菌群落的变化 (1 400~1 500)Roche 454 RDP QIIME、made4软件包、vegan软件包(PCA、相对丰度热图等)2017[49]真菌:18S rDNA葡萄发酵液西班牙歌海娜酿酒葡萄自然发酵过程中细菌和酵母群落的多样性及动态变化细菌:16S rDNA(1 540)酵母:18S rDNA(1 400~1 500)Roche 454 FLS GS、Titanium Greengenes、SILVA QIIME(Chao1指数、相对丰度热图等)2016[50]葡萄发酵液 意大利传统酿酒葡萄发酵过程中细菌群落变化情况细菌:16S rDNA V1~V3变异区(61~106)Roche 454 FLX GS、Titanium Greengenes、RDP QIIME(Chao1指数、ACE指数、Shannon指数、Simpson指数、PCA、相对丰度热图等)2015[51]葡萄发酵液 法国勃艮第葡萄酒发酵过程中酵母群落多样性 酵母:18S rDNA(1 400~1 500)Roche 454 GS GenBank、SILVA GnSPipe pipeline(Chao1指数、Shannon指数、相对丰度热图等)2014[52]
续表2

注:PCoA.主坐标分析(principal coordinates analysis);CDA.典型判别分析(canonical discriminant analysis);PLSR.偏最小二乘回归法(partial least squares regression);UPGMA.非加权组平均法(unweighted pair-group method with arithmetic mean)。
样本 研究目标 测序片段(片段长度/bp)测序平台 采用的数据库 分析软件(分析方法)发表时间葡萄发酵液南非赤霞珠葡萄发酵过程中微生物群落变化情况,并比较其与美国加州葡萄发酵过程中微生物群落间的异同真菌:ITS区(500~750)Illumina MiSeq M5RNA PAST Version 3.0、Cytoscape、Prel(Chao1指数、维恩图、Shannon指数、相对丰度热图等)2015[53]葡萄发酵液美国加州地区4 种主要葡萄品种发酵过程中微生物群落与品种、葡萄园及气候的关系细菌:16S rDNA(1 540)真菌:ITS1区(350)Illumina MiSeq Greengenes、RDP QIIME、FastTree、GenGIS II、R软件等(PCoA、CDA、PLSR等)2014[56]葡萄发酵液 发酵过程中细菌群落结构变化 细菌:16S rDNA V4~V5变异区(588~671)Illumina GAIIx 基于QIIME软件的核糖体核苷酸数据库 QIIME(PCoA、相对丰度热图等)2012[57]其他酒类安第斯吉开酒发酵液 安第斯吉开酒发酵过程中酵母菌群落多样性及其动态变化酵母:26S rDNA D1/D2区(600)Roche 454 GS Junior GenBank Mothur、R软件(Chao1指数、ACE指数、相对丰度热图、UPGMA等)2017[21]日本酒酒曲 日本酒酒曲中微生物群落组成 细菌:16S rDNA(1 540)真菌:18S rDNA ITS1区(1 750)PacBio RSll RDP QIIME(Simpson指数、Shannon指数、Chao1指数、稀释曲线、相对丰度热图、维恩图等)2017[58]大米发酵液大米发酵液分离鉴定在发酵过程中不产生物胺的微生物菌属及其在环境中的功能性发酵过程中细菌群落的动态变化情况及其与米酒挥发性风味成分之间的关系细菌:16S rDNA V4变异区(588~671)Illumina MiSeq RDP Mothur(相对丰度热图、CCA、聚类分析等)2016[54]细菌:16S rDNA V4变异区(588~671)Illumina MiSeq RDP QIIME(相对丰度热图、CCA等)2015[55]绍兴黄酒小麦曲 不同环境下黄酒小麦曲中微生物群落结构细菌:16S rDNA(1 540)Illumina HiSeq 2000 RITA CASAVA 1.8.2(Pie图、相对丰度热图等)2013[41]
从高通量测序技术诞生之日起,由于其具有测序方法简单、通量高、测序速度快和准确度较高等特点,在医学、食品、环境等研究领域受到广泛关注。随着高通量测序技术和测序平台的不断发展,该技术的精确度不断提高而成本逐渐降低,并且测序目标序列读取长度也得到提高。因此,目前高通量测序技术已被国内外研究者作为研究酿酒微生物多样性的重要手段。研究者提取不同酒样中酿酒微生物核酸后,可通过高通量测序技术直接进行测序分析。虽然测序过程简单,但其获取的测序数据信息量极为庞大,如何快速从中分析出有效的信息将成为限制研究者采用此方法分析酿酒微生物多样性的首要因素。如前文所述,数据分析方法如PCA、CCA等的使用,有助于对酿酒微生物多样性的分析,在此基础上可进一步建立起微生物多样性与功能代谢产物之间的关联[39]。
除此之外,高通量测序技术在应用中还面临着测序文库的建立等方面的挑战。在分析酿酒微生物多样性时,测序文库的建立及测序方法需依赖PCR技术。而在PCR过程中,若出现引物退火温度不适宜、模板DNA代表性不强等情况,则会在一定程度上引入系统偏差和随机性,从而导致样本中核酸分子含量与真实含量存在偏差。因此,依赖于PCR技术的高通量测序技术对样本中目标微生物定量分析的准确度有待提升[59]。建议结合使用其他不依赖于培养的技术实现目标微生物的准确鉴定和定量分析,如qPCR技术通过建立微生物浓度与PCR循环次数之间的标准曲线,可以准确进行菌群浓度的定量分析[60]。高通量测序技术若在建立测序文库时成功引入类似标准曲线,则将可以初步实现准确的定量分析。
近年来,第三代测序[61]技术逐渐发展,如DNA纳米球测序、Heliscope单分子测序、纳米孔测序、隧道电流DNA测序、质谱测序及基于显微镜测序技术等[59]。就SMRT测序技术而言,其在DNA聚合酶合成DNA分子的同时进行测序,克服了PCR带来的不确定性和偏差。与以Roche 454焦磷酸测序为代表的第二代测序技术相比,第三代测序技术在测序速度和序列长度上得到了大幅度的提升,SMRT测序技术每秒可测750 个碱基,序列长度最长可达1万 个碱基[19]。并且第三代测序平台PacBio(P4/C2)已在PacBio(XL/C2)平台的基础上大幅度提高了精确度[20]。尽管其价格昂贵,目前第三代测序技术已开始用于酿酒微生物多样性方面的分析[20]。相信随着高通量测序技术的不断完善,酿酒微生物的多样性将得到更深入、更准确、更全面的分析与研究。
[1] XU Y, WANG D, FAN W L, et al. Traditional Chinese biotechnology[J].Advances in Biochemical Engineering/Biotechnology,2009, 122: 189-233.doi:10.1007/10_2008_36.
[2] JIN G Y, ZHU Y, XU Y. Mystery behind Chinese liquor fermentation[J]. Trends in Food Science and Technology, 2017, 63:18-28.doi:10.1016/j.tifs.2017.02.016.
[3] 傅金泉. 中国近代酿酒微生物研究史料[J]. 酿酒科技, 2006(5): 82-88.
[4] WANG C X, ESTEVE-ZARZOSO B, MAS A. Monitoring of Saccharomyces cerevisiae, Hanseniaspora uvarum, and Starmerella bacillaris (synonym Candida zemplinina) populations during alcoholic fermentation by fluorescence in situ hybridization[J]. International Journal of Food Microbiology, 2014, 191: 1-9.doi:10.1016/j.ijfoodmicro.2014.08.014.
[5] HIERRO N, ESTEVE-ZARZOSO B, GONZÁLEZ Á, et al. Realtime quantitative PCR (QPCR) and reverse transcription-QPCR for detection and enumeration of total yeasts in wine[J]. Applied and Environmental Microbiology, 2006, 72(11): 7148-7155.doi:10.1128/AEM.00388-06.
[6] ANDORRA I, LANDI S, MAS A, et al. Effect of fermentation temperature on microbial population evolution using cultureindependent and dependent techniques[J]. Food Research International, 2010, 43(3): 773-779.doi:10.1016/j.foofres.2009.11.014.
[7] HAZEN T C, DUBINSKY E A, DESANTIS T Z, et al. Deep-sea oil plume enriches indigenous oil-degrading bacteria[J]. Science, 2010,330: 204-208.doi:10.1126/science.1195979.
[8] FIERER N, LAUBER C L, ZHOU N, et al. Forensic identification using skin bacterial communities[J]. Proceedings of the National Academy of Sciences, 2010, 107(14): 6477-6481.doi:10.1073/pnas.100162107.
[9] SHENDURE J, JI H. Next-generation DNA sequencing[J]. Nature Biotechnology, 2008, 26(10): 1135-1145.doi:10.1038/nbt1486.
[10] WANG Chunxiao, GARCÍA-FEMÁNDEZ D, MAS A, et al.Fungal diversity in grape must and wine fermentation assessed by massive sequencing, quantitative PCR and DGGE[J]. Frontiers in Microbiology, 2015, 6: 1156.doi:10.3389/fmicb.2015.01156.
[11] WANG C X, ESTEVE-ZARZOSO B, COCOLIN L, et al. Viable and culturable populations of Saccharomyces cerevisiae, Hanseniaspora uvarum and Starmerella bacillaris (synonynm Candida zemplinina)during Barbera must fermentation[J]. Food Research International,2016, 78: 195-200.doi:10.1016/j.foodres.2015.10.014.
[12] SUN W N, XIAO H Z, PENG Q, et al. Analysis of bacterial diversity of Chinese Luzhou-flavor liquor brewed in different seasons by Illumina MiSeq sequencing[J]. Annals of Microbiology, 2016, 66(3): 1293-1301.doi:10.1007/s13213-016-1223-5.
[13] ZHANG Xiuhong, ZHAO Jinglong, DU Xiaowei. Barcoded pyrosequencing analysis of the bacterial community of Daqu for lightflavour Chinese liquor[J]. Letters in Applied Microbiology, 2014, 58(6): 549-555.doi:10.1111/lam.12225.
[14] WANG L, WANG Y Y, WANG D Q, et al. Dynamic changes in the bacterial community in Moutai liquor fermentation process characterized by deep sequencing[J]. Journal of the Institute of Brewing, 2015, 121(4): 603-608.doi:10.1002/jib.259.
[15] QUAST C, PRUESSE E, YILMAZ P, et al. The SILVA ribosomal RNA gene database project: improved data processing and webbased tools[J]. Nucleic Acids Research, 2013, 41(D1): D590-D596.doi:10.1093/nar/gks1219.
[16] LIU L, LI Y H, LI S L, et al. Comparison of next-generation sequencing systems[J]. Journal of Biomedicine and Biotechnology, 2012(7): 251364.doi:10.1155/2012/251364.
[17] QUAIL M A, SMITH M, COUPLAND P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacif i c biosciences and Illumina MiSeq sequencers[J]. BMC Genomics, 2012, 13(1): 341.doi:10.1186/1471-2164-13-341.
[18] KORLACH J, BJORNSON K P, CHAUDHURI B P, et al. Realtime DNA sequencing from single polymerase molecules[J]. Methods in Enzymology, 2010, 472: 431-455.doi:10.1016/S0076-6870(10)72001-2.
[19] OZOLAK F. Third-generation sequencing techniques and applications to drug discovery[J]. Expert Opinion on Drug Discovery, 2012, 7(3):231-243.doi:10.1517/17460441.2012.660145.
[20] MOSHER J J, BOWRNAN B, BEMBERG E L, et al. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing[J]. Journal of Microbiological Methods, 2014, 104: 59-60. doi:10.1016/j.mimet.2014.06.012.
[21] MENDOZA L M, NEEF A, VIGNOLO G, et al. Yeast diversity during the fermentation of Andean chicha: a comparison of high-throughput sequencing and culture-dependent approaches[J]. Food Microbiology, 2017, 67: 1-10.doi:10.1016/j.fm.2017.05.007.
[22] COLE J R, WANG Q, CARDENAS E, et al. The ribosomal database project: improved alignments and new tools for rRNA analysis[J].Nucleic Acids Research, 2009, 37: D141-D145.doi:10.1093/nar/gkn879.
[23] DESANTIS T Z, HUGENHOLTZ P, LARSEN N, et al. Greengenes:chimera-checked 16S rRNA gene database and work bench compatible in ARB[J]. Applied and Environmental Microbiology, 2006, 72(7):5069-5072.doi:10.1128/AEM.03006-05.
[24] ABARENKOV K, HENRIK N R, LARSSON K H, et al. The UNITE database for molecular identification of fungi-recent updates and future perspectives[J]. New Phytologist, 2010, 186(2): 281-285.doi:10.1111/j.1469-8137.2009.03160.x.
[25] SCHLOSS P D, WESTCOTT S L, RYABIN T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities[J].Applied and Environmental Microbiology, 2009, 75(23): 7537-7541.doi:10.1128/AME.01541-09.
[26] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 2010, 7(5): 335-336.doi:10.1038/nmeth.f.303.
[27] 赵亮, 王莉, 汪地强, 等. 白酒微生物群落研究技术现状与二代测序数据分析方略[J]. 酿酒科技, 2016(7): 88-96.
[28] 孙宝国, 孙金沅, 宫俐莉, 等. 中国白酒中长期发展趋势与研究重点之管见[J]. 轻工学报, 2016, 31(1): 6-11.
[29] XU M L, YU Y, RAMASWAMY H S, et al. Characterization of Chinese liquor aroma components during aging process and liquor age discrimination using gas chromatography combined with multivariable statistics[J]. Scientific Reports, 2017, 7: 39671.doi:10.1038/srep39671.
[30] PARDO-MATES N, VERA A, BARBOSA S, et al. Characterization classification and authentication of fruit-based extracts by means of HPLC-UV chromatographic fingerprints, polyphenolic profiles and chemometric methods[J]. Food Chemistry, 2017, 221: 29-38.doi:10.1016/j.foodchem.2016.10.033.
[31] WANG X, HU X Y, DENG K, et al. High-throughput sequencing of microbial diversity in implant-associated infection[J]. Infection Genetics and Evolution, 2016, 43: 307-311.doi:10.1016/j.meegid.2016.06.006.
[32] SERRANO-LOURIDO D, SAURINA J, HERNÁDEZ-CASSOU S, et al.Classification and characterisation of Spanish red wines according to their appellation of origin based on chromatographic pro fi les and chemometric data analysis[J]. Food Chemistry, 2012, 135(3): 1425-1431.doi:10.1016/j.foodchem.2012.06.010.
[33] WANG X D, BAN S D, HU B D, et al. Bacterial diversity of Moutaifl avour Daqu base on high-throughput sequencing method[J]. Journal of the Institute of Brewing, 2017, 123(1): 138-143.doi:10.1002/jib.391.
[34] ZHENG Xiaowei, YAN Zheng, NOUT M J R, et al. Microbiota dynamics related to environmental conditions during the fermentative production of Fen-Daqu, a Chinese industrial fermentation starter[J]. International Journal of Food Microbiology, 2015, 182/183(28): 57-62.doi:10.1016/j.ijfoodmicro.2014.05.008.
[35] XI B D, ZHAO X Y, HE X S, et al. Successions and diversity of humic-reducing microorganisms and their association with physicalchemical parameters during composting[J]. Bioresource Technology,2016, 219: 204-211.doi:10.1016/j.biortech.2016.07.120.
[36] TANG Jing, TANG Xiaoxin, TANG Ming, et al. Analysis of the bacterial communities in two liquors of soy sauce aroma as revealed by high-throughput sequencing of the 16S rRNA V4 hypervariable region[J]. BioMed Research International, 2017, 2017: 1-9.doi:10.1155/2017/6271358.
[37] 郭敏, 黄永光, 邱树毅, 等. 高通量测序在酱香白酒微生态多样性研究中的应用[J]. 中国酿造, 2017, 36(5): 146-151.doi:10.11882/j.issn.0254-5071.2017.05.031.
[38] 黄蕴利, 黄永光, 胡建峰, 等. 酱香型白酒第二轮次酒发酵过程微生物多样性研究[J]. 中国酿造, 2017, 36(9): 30-35.doi:10.11882/j.issn.0254-5071.2017.09.007.
[39] ZHENG Qi, LIN Bairong, WANG Yibin, et al. Proteomic and highthroughput analysis of protein expression and microbial diversity of microbes from 30- and 300-year pit muds of Chinese Luzhouflavor liquor[J]. Food Research International, 2015, 75: 305-314. doi:10.1016/j.foodres.2015.06.029.
[40] PORTILLO M D C, FRANQUÈS J, ARAQUE I, et al. Bacterial diversity of Grenache and Carignan grape surface from different vineyard at Priorat wine region (Catalonia, Spain)[J]. International Journal of Food Microbiology, 2016, 219: 56-63.doi:10.1016/j.ijfoodmicro.2015.12.002.
[41] XIE Guangfa, WANG Lan, GAO Qikang, et al. Microbial community structure in fermentation process of Shaoxing rice wine by Illuminabased metagenomic sequencing[J]. Journal of the Science of Food and Agriculture, 2013, 93(12): 3121-3125.doi:10.1002/jsfa.6058.
[42] BOKULICH N A, MILLS D A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-highthroughput prof i ling of fungal communities[J]. Applied Environmental Microbiology, 2013, 79(8): 2519-2526.doi:10.1128/AME.03870-12.
[43] DAVID V, TERRAT S, HERZINE K, et al. High-throughput sequencing of amplicons for monitoring yeast biodiversity in must and during alcoholic fermentation[J]. Journal of Industrial Microbiology and Biotechnology,2014, 41(5): 811-821.doi:10.1007/s10295-014-1427-2.
[44] PINTO C, PINTO D, CARDOSO R, et al. Wine fermentation microbiome a landscape from different Portuguese wine appellations[J]. Frontiers in Microbiology, 2015, 6: 905.doi:10.3389/fmicb.2015.00905.
[45] WANG X S, DU H, XU Y. Source tracking of prokaryotic communities in fermented grain of Chinese strong-flavor liquor[J]. International Journal of Food Microbiology, 2017, 244: 27-35.doi:10.1016/j.ijfoodmicro.2016.12.018.
[46] HU X, DU H, REN C, et al. Illuminating anaerobic microbial community and co-occurrence patterns across a quality gradient in Chinese liquor fermentation pit muds[J]. Applied & Environmental Microbiology, 2016, 82(8): 2506-2515.doi:10.1128/AME.03409-15.
[47] 陈玲, 袁玉菊, 曾丽云, 等. 16S rDNA克隆文库法与高通量测序法在浓香型大曲微生物群落结构分析中的对比研究[J]. 酿酒科技,2015(12): 33-36; 40.
[48] 周森, 胡佳音, 赵卫鹏, 等. 高通量测序技术在航天大曲微生物多样性中的应用[J]. 酿酒科技, 2016(9): 76-78.doi:10.13746/j.njj.2016122.
[49] DE FILIPPIS F, LA STORIA A, BLAOPTTA G. Monitoring the mycobiota during Greco di Tufo and Aglianico wine fermentation by 18S rRNA gene sequencing[J]. Food Microbiology, 2017, 63: 117-122. doi:10.1016/j.fm.2016.11.010.
[50] PORTILLO M D C, MAS A. Analysis of microbial diversity and dynamics during wine fermentation of Grenache grape variety by high-throughput barcoding sequencing[J]. LWT-Food Science and Technology, 2016, 72: 317-321.doi:10.1016/j.lwt.2016.05.009.
[51] PIAO H, HAWLEY E, KOPF S, et al. Insights into the bacterial community and its temporal succession during the fermentation of wine grapes[J]. Frontiers in Microbiology, 2015, 6: 809.doi:10.3389/fmicb.2015.00809.
[52] DAVID V, TERRAT S, HERZINE K, et al. High-throughput sequencing of amplicons for monitoring yeast biodiversity in must and during alcoholic fermentation[J]. Journal of Industrial Microbiology and Biotechnology, 2014, 41(5): 811-821.
[53] SETATI M E, JACOBSON D, BAUER F F. Sequence-based analysis of the Vitis vinifera L. cv Cabernet Sauvignon grape must mycobiome in three south African vineyards employing distinct agronomic system[J]. Frontiers in Microbiology, 2015, 6(6): 1358.doi:10.3389/fmicb.2015.03158.
[54] LIU S P, YU J X, WEI X L, et al. Sequencing-based screening of functional microorganism to decrease the formation of biogenic amines in Chinese rice wine[J]. Food Control, 2016, 64: 98-104.doi:10.1016/j.foodcont.2015.12.013.
[55] LIU S P, MAO J, LIU Y Y, et al. Bacterial succession and the dynamics of volatile compounds during the fermentation of Chinese rice wine from Shaoxing region[J]. World Journal of Microbiology and Biotechnology, 2015, 31(12): 1907-1921.doi:10.1007/s11274-015-1931-1.
[56] BOKULICH N A, THOMGATE J A, RICHARDSON P M, et al.Microbial biogeography of wine grapes in conditioned by cultivar,vintage, and climate[J]. Proceeding of the National Academy of Sciences, 2014, 111(1): 139-148.doi:10.1073/pnas.1317377110.
[57] BOKULICH N A, JOSEPH C M L, ALLEN G, et al. Next-generation sequencing reveals signif i cant bacterial diversity of botrytized wine[J]. PLoS ONE, 2012, 7(5): e36357.doi:10.1371/journal.phone.0036357.
[58] HUI W Y, HOU Q C, CAO C X, et al. Identification of microbial profile of koji using single molecule, real-time sequencing technology[J]. Journal of Food Science, 2017, 82(5): 1193-1199.doi:10.1111/1750-3841.13699.
[59] MAYO B, RACHID C, ALEGRÍA Á, et al. Impact of next generation sequencing techniques in food microbiology[J]. Current Genomics, 2014, 15(4): 293-309.doi:10.2174/1389202915666140616233211.
[60] BELDA I, ZARRAONAINDIA I, PERISIN M, et al. From vineyard soil to wine fermentation: microbiome approximations to explain the “terroir” concept[J]. Frontiers in Microbiology, 2017, 8: 821.doi:10.3389/fmicb.2017.00821.
[61] SCHADT E E, TURNER S, KASARSKIS A. A window into thirdgeneration sequencing[J]. Human Molecular Genetics, 2010, 19(R2): 227-240.doi:10.1093/hmg/ddq481.
A Review of the Application of High-Throughput Sequencing Technology in Analysis of the Diversity of Microbial Communities Involved in the Fermentation of Alcoholic Beverages
第一作者简介:吴成(1993—)(ORCID: 0000-0002-0385-3100),男,硕士研究生,研究方向为发酵工程。E mail: 742990507@qq.com
mail: 742990507@qq.com
王春晓(1987—)(ORCID: 0000-0001-8646-863X),女,讲师,博士,研究方向为酿酒工程、食品生物技术。E mail: cxwang@gzu.edu.cn
mail: cxwang@gzu.edu.cn
邱树毅(1963—)(ORCID: 0000-0001-7588-0573),男,教授,博士,研究方向为酿酒工程、食品生物技术。E mail: syqiu@gzu.edu.cn
mail: syqiu@gzu.edu.cn