海洋氧化节杆菌KQ11右旋糖苷酶催化位点关键氨基酸
刘 乐1,丁 一1,王紫玄1,房耀维1,王淑军1,2,吕明生1,*
(1.淮海工学院海洋生命与水产学院,江苏 连云港 222005;2.江苏省海洋资源开发研究院,江苏 连云港 222005)
摘 要: 通过同源比对,对来自海洋氧化节杆菌(Arthrobacter oxydans KQ11)的右旋糖苷酶(记作AoDex)催化域及关键氨基酸进行预测,运用定点突变将AoDex催化域418-QTDGIELYKGSTMKNTFFNANDD-440中的5 个氨基酸突变为甘氨酸,获得5 种突变型右旋糖苷酶原核表达载体:pColdIII-KQN-Q418G、pColdIII-KQN-D420G、pColdIII-KQN-E423G、pColdIII-KQN-D439G、pColdIII-KQN-D440G,表达产物分别记为Q418GDex、D420GDex、E423GDex、D439GDex、D440GDex。经过发酵表达,Q418GDex、D420GDex、E423GDex、D439GDex几乎没有酶活力。D440GDex酶活力与AoDex一致;所不同的是,D440GDex在25~40 ℃时的酶活力提高了2~3 倍,最适pH值也从AoDex的5.5变为6.5。数据表明,Q418、D420、E423、D439四个氨基酸残基是AoDex催化域中的关键氨基酸。D440突变为甘氨酸对该酶的性质有较大影响,也表明其不是催化域中的广义碱。本研究表明AoDex的催化机制与糖苷酶49家族是一致的,为AoDex的功能改造提供了理论支持。
关键词: 右旋糖苷酶;定点突变;催化域;关键氨基酸;结构预测
右旋糖苷酶(EC.3.2.1.11)水解α-1,6糖苷键产生异麦芽寡糖,广泛应用于食品、化工和医药行业[1-5]。在制糖工业中,该酶被用于降低甘蔗汁黏度、减少管道堵塞、提高糖产量和品质[6-10]。在化工领域,该酶被用于合成色谱填料、增稠剂、乳化剂等[11]。在口腔保健医学中,该酶用于清除牙菌斑,预防龋齿、牙结石和牙周病[1]。在医药行业,该酶可用于生产代血浆的低分子质量右旋糖酐。我国2012年批准右旋糖苷酶为食品添加剂,其在食品加工中应用将越来越广泛。
本实验室从海泥中筛选出1 株产右旋糖苷酶的氧化节杆菌KQ11(Arthrobacter oxydans KQ11)[12-13],对右旋糖苷酶学性质进行了研究,构建了该酶的基因工程菌[14-17]。目前,在蛋白质数据库(protein data base,PDB)中已解析结构的右旋糖苷酶仅有:Penicillium minioluteum产的右旋糖苷酶(PmDex),包含氨基酸残基574 个[18];Streptococcus mutans产的右旋糖苷酶(SmDex),包含氨基酸残基643 个[19];Thermoanaerobacter pseudethanolicus产的右旋糖苷酶(TpDex),包含氨基酸残基1 236 个[20]。它们分属糖苷酶49和66家族[21-22],催化机制分别为构型翻转和构型保持[18-19]。相比其他糖苷酶,右旋糖苷酶的结构与功能信息过少。氧化节杆菌右旋糖苷酶(AoDex)催化域的研究鲜见报道。
通过生物信息学方法对酶蛋白结构解析,定点突变催化域中的保守氨基酸残基,是对催化域关键氨基酸研究的主要方法[20]。右旋糖苷酶在食品加工中应用受到其酶学性质的限制。制糖行业要求酶活力高、耐高温、稳定性好的右旋糖苷酶[6]。在代血浆右旋糖酐生产中,需要控制产物分子质量和催化方向,通过对酶进行分子改造可以实现。本研究通过同源比对和结构预测以及对关键氨基酸定点突变,研究AoDex的催化域及其关键氨基酸残基,为该酶的结构解析和进一步的结构改造提供理论支持。
1 材料与方法
1.1 材料与试剂
菌株为本实验室构建并保藏,该菌株的宿主菌为大肠杆菌(Escherichia coli)BL21(DE3),质粒为pColdIII,目的基因为来自KQ11的1 923 bp右旋糖苷酶基因。
感受态细胞E. coli DH5α和E. coli BL21(DE3)杭州宝赛生物科技有限公司;高纯度质粒小提试剂盒杭州宝赛生物科技有限公司;快速定点突变试剂盒天根生化科技(北京)有限公司;HisPur Ni-NAT Superflow Agarose 美国Thermo Fisher公司;密理博超滤管 美国密理博公司;异丙基硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG) 美国Sigma公司;引物合成及DNA测序由生工生物工程(上海)股份有限公司完成。
1.2 仪器与设备
PowerPac™ HC电泳仪 美国Bio-Rad公司;DG-3A微型水平凝胶电泳槽 北京鼎国生物技术有限公司;Multiskan GO全波长酶标仪 美国Thermo Fisher公司;PyxisTM凝胶分析系统 南京思普金生物科技有限公司;Q5000微量分光光度计 美国Quawell公司;JY92-IIN超声细胞破碎仪 宁波新芝生物科技股份有限公司。
1.3 方法
1.3.1 重组质粒提取
E. coli KQ11-ColdIII在LB培养基(蛋白胨1%,酵母粉0.5%,氯化钠1%,50 μg/mL氨苄,pH 7.5),37 ℃、180 r/min培养过夜,用高纯度质粒小提试剂盒提取质粒。
1.3.2 右旋糖苷酶催化域关键氨基酸的选择
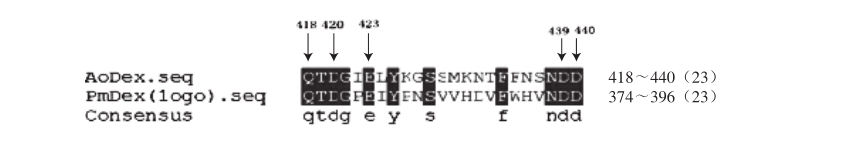
AoDex氨基酸序列提交JPred在线服务器(http://www.compbio.dundee.ac.uk/jpred/),通过对数据库的搜索,发现该序列与朱黄青霉(P. minioluteum)(PDB code:1OGO)[18]右旋糖苷酶(PmDex)和黑曲酶(Aspergillus niger)ATCC9642所产的异普鲁兰酶(PDB code:2Z8G)[22]的序列最为相近,同属GH-49,酶蛋白表面的缝隙中的氨基酸残基构成了催化域。对AoDex氨基酸序列与PmDex氨基酸序列进行比对分析。
根据PDB和碳水化合物活性酶维基(carbohydrate active enzymes pedia,CAZy)数据库的记载,PmDex催化机制为构型翻转,催化区域为Q374~D396。选择与PmDex催化区域内保守一致的氨基酸作为AoDex的突变位点。为便于突变酶之间的比较,故将突变位点氨基酸都突变为甘氨酸。
1.3.3 定点突变
用快速定点突变试剂盒以未突变的重组质粒为模板进行聚合酶链式反应(polymerase chain reaction,PCR),引物设计见表1。反应体系(50 μL):Fast Alteration DNA聚合酶(2.5 U/μL)1.5 μL;5×FastAlteration Buffer 10 μL;未突变质粒模板2 μL;正向引物(10 μmol/L)2 μL;反向引物(10 μmol/L)2 μL;ddH2O 32.5 μL。反应条件:95 ℃预变性2 min;94 ℃变性20 s;55 ℃退火10 s;68 ℃延伸4 min;共18 个循环;68 ℃延伸5 min;10 ℃保存。50 μL PCR产物加入1 μLDpnI在37 ℃孵育1 h,消化模板。吸取10 μL消化产物加入到100 μL感受态细胞E. coliDH5α,冰浴30 min,42 ℃准确热激90 s,冰浴2 min。加入700 μL无抗LB培养基,37 ℃、200 r/min培养1 h。吸取300 μL培养后的混合物均匀涂布在含氨苄抗性的LB平板上,37 ℃培养过夜。挑取阳性菌落培养用于后续实验。
表1 定点突变引物
Table 1 Primer sequences used for site-directed mutation

注:下划线表示突变位点。
引物名称 碱基序列(5′-3′) 突变酶F-Q418G TACAGTTCGATGCCGTCGGTGCCCCAGTACCAGCTGCCCACCT Q418GDex R-Q418G CCCTTGTACAGTTCGATGCCGGCCGTCTGCCAGTACCAGCTGC D420GDex R-D420G AGGTGGGCAGCTGGTACTGGGGCACCGACGGCATCGAACTGTA F-D420G GCAGCTGGTACTGGCAGACCGGCGGCATCGAACTGTACAAGGG F-E423G AGACCGACGGCATCGGCCTGTACAAGGGC E423GDex R-E423G TGGTACATCTTCAGCACGTCGGCGTTCGCG TTGAAGAAGGTGT D439GDex R-D439G GCCGATGCCGTCGGTCTGCCAGTACCAG F-D439G CCGTCGTTCGCGTTGAAGAAGGTGTTC TCAACGCGAACGACGGCGTGCTGAAGAT D440GDex R-D440G ACACCTTCTTCAACGCGAACGCCGACGTGCTGAAGATGTACCA F-D440G
1.3.4 发酵产酶与纯化
将含有突变质粒的E. coli BL21(DE3)接种到含50 μg/mL的氨苄青霉素的LB培养基,37 ℃、180 r/min培养5.5 h活化,5%接种含氨苄LB培养基,37 ℃、180 r/min培养至OD值0.8左右(15 h左右)。加入IPTG使其终浓度为0.5 mmol/L,20 ℃诱导48 h。菌悬液12 396×g离心20 min,沉淀用20 mmol/L pH 7.5的Tris-HCl溶液1 mL重悬,超声破碎6 min,12 396×g离心20 min,收集上清液。用Ni-NAT柱对发酵上清液进行纯化,将咪唑洗脱缓冲液用截留分子质量为10 kDa的密理博超滤管进行浓缩,检测蛋白浓度,进行15%的十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfatepolyacrylamide gelelectrophoresis,SDS-PAGE),检测酶活力。
1.3.5 右旋糖苷酶活力测定
3,5-二硝基水杨酸(3,5-dinitrosalicylic acid,DNS)法进行酶活力测定[15],取50 μL酶液于10 mL试管中,加入150 μL 3%的右旋糖苷酐乙酸乙酸钠缓冲液(50 mmol/L,pH 5.5),在45 ℃水浴中反应15 min,冷却,加入200 μL DNS终止反应,沸水中显色5 min。加入3 mL蒸馏水,混匀,测定540 nm吸光度,数值代入麦芽糖标准曲线方程计算酶活力。酶活力单位定义:在pH 5.5、45 ℃条件下,每分钟催化产生1 μmol麦芽糖所需的酶量,即为1 个酶活力单位(U)。
1.3.6 突变酶的酶作用温度和pH值的测定
将酶分别置于25~70 ℃的水浴锅中测定酶活力。酶作用pH值研究首先配制50 mmol/L乙酸-乙酸钠缓冲液(pH 4.0、4.5、5.0、5.5、6.0、6.5),50 mmol/L Britton-Robinson缓冲溶液(pH 6.5、7.0、7.5、8.0),然后用不同的缓冲液配制含3%的右旋糖酐底物溶液,在45 ℃水浴中测定不同pH值条件下的酶活力。
1.3.7 结构分析与同源建模
采用Expasy提供的Prot Param程序对右旋糖苷酶的氨基酸残基数目、组成、原子组成、蛋白相对分子质量、理论等电点及疏水性平均值等参数进行在线分析。
采用SOPMA对该蛋白的二级结构进行预测分析。
使用SWISS-MODEL对该蛋白的三维结构进行预测,并利用Procheck和Verify_3D对蛋白质结构模型进行评估。使用PyMol2.7软件展示三级结构模型。
1.4 数据及图像处理
序列比对与使用DNAMAN 6.0处理并展示,数据处理使用Origin 2017,氨基酸相互作用处理使用LigPlus。
2 结果与分析
2.1 催化位点的预测结果
AoDex与PmDex的氨基酸序列比对结果见图1,AoDex的Q418~D440与PmDex的催化区域Q374~D396中的催化位点具有高度保守性(47.83%的氨基酸序列一致性,图2)。PmDex催化区域的3 个天冬氨酸残基D376、D395和D396是该糖苷酶家族催化域的保守残基。AoDex的氨基酸序列的Q418、T419、D420、G421、E423、Y425、S428、F434、N438、D439与PmDeX催化区域的氨基酸残基一致。与PmDex的Q374对应的AoDex的Q418,PmDex的D376、D395和D396对应AoDex的D420、D439和D440推断为催化氨基酸。在糖苷酶中,天冬氨酸和谷氨酸通常都可为催化域中的广义酸碱对,因此推断E423也有可能参与催化反应。
2.2 定点突变检测结果
定点突变后的质粒转化入感受态E. coliDH5α,37 ℃培养过夜。提取质粒,经生工生物工程(上海)股份有限公司测序,5 个选定的突变位点都成功地突变为设计的碱基。在定点突变的引物设计参考了不同文献[20],Q418、D420、E439是把突变位点放在引物中间,而E423、D440的引物则是突变位点偏向一边。在定点突变试验中,如试验多次没有达到预期结果时,可通过改变引物设计策略,以确保实验成功。
2.3 蛋白表达检测结果
为检测突变后的基因是否正常表达,对5 个突变型37 ℃发酵,IPTG诱导表达,产物经Ni-NAT柱纯化,使用SDS-PAGE检测。如图3所示,表达蛋白的分子质量在66 kDa左右,与AoDex的分子质量是一致的,5 个定点突变菌株全部成功表达了右旋糖苷酶蛋白。
2.4 突变型与未突变型右旋糖苷酶活力的测定结果
如图4所示,检测的Q 4 1 8 G D e x酶活力为0.021 U/mL,D420GDex酶活力为0.158 U/mL,E423GDex酶活力为0.060 U/mL,D439GDex酶活力为0.078 U/mL,与AoDex相比几乎没有酶活力。检测到的D440GDex酶活力为50 U/mL,与野生型酶活力相比是一致的。因此,Q418、D420、E423和D439为AoDex催化域的关键氨基酸。当分别被替换为甘氨酸时,失去了催化活性。D440虽然紧邻D439,在其被替换为甘氨酸时,并没有影响到该酶的催化活性。在PmDex的CAZy数据中,D376和D396中的1 个或2 个都是广泛碱。D440与PmDex催化域中的保守残基D396是一致的,本试验表明D440并不是催化域中的广义碱,由此推断,PmDex中的D396可能也不是其催化域中的广义碱。
2.5 温度和pH值对突变右旋糖苷酶的影响
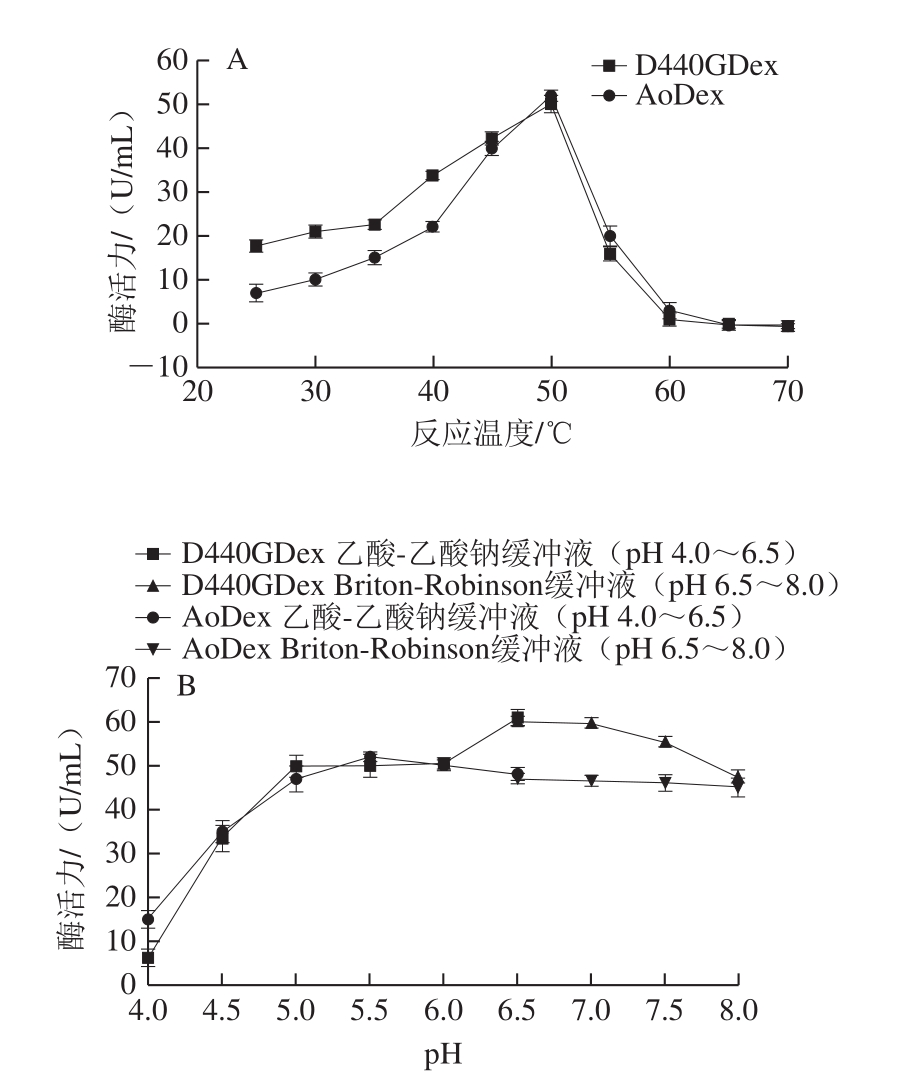
如图5A所示,D440GDex的最适酶促反应温度与突变前保持一致,然而,当反应温度在25~40 ℃时,酶的活力显著提高。突变前,在此温度间的酶活力分别为5.5、9.5、14.5、20.5 U/mL;突变后,分别为17.7、21.6、33.0、33.8 U/mL。D440GDex活力分别是AoDex活力的321%、227%、228%和165%。D440紧邻催化区域,对底物右旋糖苷酐与酶的接合是有影响的。温度影响到分子动力学,对于酶结合位点与催化位点的结构影响较大[23-34]。定点突变把天冬氨酸变为甘氨酸,在结构上少了一个带负电的乙酸,扩大的催化域边缘处的空间,降低了静电作用。这可能是突变酶在相对低的温度下,仍能与底物顺畅的结合。而随着温度的逐步升高,这种优势逐渐减小。
从最适pH值的角度(图5B),AoDex的最适作用pH值为5.5,此时,酶活力为52 U/mL;而D440GDex的最适作用pH值为6.5,酶活力达到61 U/mL,酶活力提高了20%。pH值的改变将导致底物和酶分子的带电状态发生变化。D440GDex在pH 6.5~7.5时的酶活力都高于AoDex。D440的突变使催化域边缘少了一个羰基,蛋白质的等电点向碱性方向偏移[25],经计算,AoDex的等电点为5.19,而D440GDex等电点变为5.23。对催化域局部而言,减少了1 个离子键或氢键作用对其自身或底物的作用。在pH 6.5~7.5之间,D440GDex的催化域结构没有受到影响。D440GDex在25~40℃酶活力的大幅提高以及最适pH值偏碱都有利于其在口腔护理产品中的应用[17]。
2.6 AoDex及突变体的结构分析
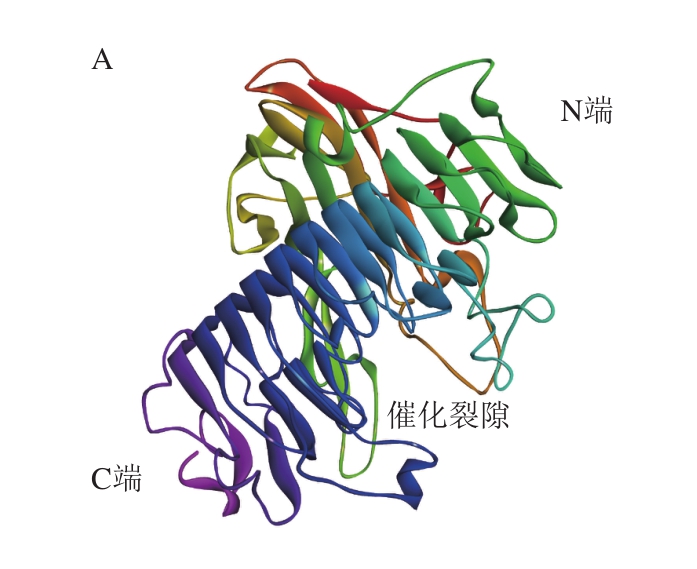
通过Prot Param程序分析显示,AoDex由641 个氨基酸组成,使用SWISS-MODLE进行三维同源建模可以得到2 个AoDex三维模型,利用Chrion对模型进行精修,减少结构的能量排斥,选择总体能量最小的模型作为基本模型(图6A)。用Procheck检测优化后模型的立体化学合理性,蛋白的φ、ψ二面角81%在核心区域,16.7%在额外允许区,1.2%处于最大允许区,仅有1.2%在不可信区域(图6B)。Verify Protein(Profiles-3D)没有检测到不合理Loop区域。利用Easymodeller 2.0对得到的模型进行优化。对优化后的模型用Errat进行评估得分85.940(图6C)。由此得到的三维模型与PmDex的三维模型相似。
图6A显示AoDex有2 个结构域,一个位于N端的β-夹层结构,另一个是右手平行的β-螺旋结构。在后者的分子表面存在一个裂隙。预测的催化位点即位于该缝隙处,见图6D,Q418、D420、E423、D439和D440的R基均为羧基且暴露在缝隙处外侧,经过突变为甘氨酸,羧基变为H,失去了广义酸碱提供或是接受H+的作用[26-27],也就失去了催化作用。依据同源比对的PmDex的催化域,AoDex的D439应为广义酸,其将与底物+1亚位点的糖苷氧表成一个氢键,D420则为广义碱,其活化水分子,该水分子再亲和攻击右旋糖苷α-1,6糖苷键上的异头碳,实现糖苷键的断裂[28]。
突变体Q418GDex和E423GDex酶活力检测几乎为零。在PmDex的催化域中也是保守的。谷氨酰胺残基和谷氨酸残基虽然不参与催化反应,然而,因减少了离子键或是氢键的作用,造成催化域局部构象发生改变。使用LigPlus对氨基酸的相互作用进行分析。如图7A所示,未突变前Gln418能够分别与Tyr342和Glu343形成氢键,且距离分别为2.93 Å和3.02 Å,Thr419分别与Gln411、Ser414、Tyr416和Asn438形成氢键,距离分别3.11、3.11、2.78 Å和2.99 Å;图7B显示突变为Gly418后,Gly418分别与His391、Thr419、Asp439形成氢键,距离分别为2.91、2.88、3.32 Å,Thr419与Gln411、Ser414、Tyr416的距离分别改变3.16、3.03、2.76 Å。图7C显示未突变前Glu423能够与Lys443形成氢键,之间的距离为2.65 Å;图7D显示突变为Gly423后,氢键作用消失。催化域中广义酸碱之间距离改变[29-30],攻击糖苷键的能力丧失。当然,AoDex催化域结构中各个残基的作用,最终还需要通过晶体与配体的结构解析才能确定[25]。
3 结 论
对来自海洋氧化节杆菌(A. oxydans KQ11)的右旋糖苷酶(AoDex)的催化域及关键氨基酸进行预测,运用定点突变将AoDex催化域中Q418、D420、E423、D439、D440氨基酸变为甘氨酸,对5 种突变型右旋糖苷酶原核表达,Q418GDex、D420GDex、E423GDex、D439GDex几乎没有酶活力。D440GDex酶活力为50 U/mL,与AoDex一致。D440GDex在25~40 ℃时的酶活力比突变前提高了2~3 倍;最适pH值也从AoDex的5.5变为6.5。数据表明,Q418、D420、E423、D439四个氨基酸残基是AoDex催化域中的关键氨基酸。D440的突变对该酶的性质有较大影响,也表明其不是催化域中的广义碱。本研究为探索AoDex的结构与功能改造提供一定的参考。
参考文献:
[1] 吴敏, 张宇馨, 张洪斌, 等. 右旋糖酐酶的克隆表达、性质表征及其应用研究[C]//中国酶工程学术研讨会, 2017.
[2] JMR T, GOMES W F, RODRIGUES S. Efficient production of prebiotic gluco-oligosaccharides in orange juice using immobilized and co-immobilized dextransucrase[J]. Applied Biochemistry &Biotechnology, 2017, 183(4): 1-17. DOI:10.1007/s12010-017-2496-2.
[3] SANTOS M, TEIXEIRA J, RODRIGUES A. Production of dextransucrase, dextran and fructose from sucrose using Leuconostoc mesenteroides NRRLB512(F)[J]. Biochemical Engineering Journal,2000, 4(3): 177-188. DOI:10.1016/S1369-703X(99)00047-9.
[4] ŁUKASZEK M. The Problem of dimorphism in cryptophytespreliminary studies on the occurrence of Cryptomonas spp. in chosen water bodies[C]//Wpływ Młodych Naukowców na Osiągnięcia Polskiej Nauki-VII Edycja, 2014.
[5] WANG X, CHENG H, LU M, et al. Dextranase from Arthrobacter oxydans KQ11-1 inhibits biofilm formation by polysaccharide hydrolysis[J]. Biofouling, 2016, 32(10): 1223-1233. DOI:10.1080/089 27014.2016.1239722.
[6] JIMÉNEZ E R. Dextranase in sugar industry: a review[J]. Sugar Tech,2009, 11(2): 124-134. DOI:10.1007/s12355-009-0019-3.
[7] ZHANG Z D, LIU J D, MA S Y, et al. Enhancement of catalytic performance of α-dextranase from Chaetomium gracile, through optimization and suitable shear force[J]. Sugar Tech, 2017, 20(1): 78-87: 1-10. DOI:10.1007/s12355-017-0540-8.
[8] LI K, LU H Q, HANG F X, et al. Improved dextranase production by Chaetomium gracile, through optimization of carbon source and fermentation parameters[J]. Sugar Tech, 2017, 19(4): 432-437.DOI:10.1007/s12355-016-0476-4.
[9] EGGLESTON G, KARR J, PARRIS A, et al. A rapid biochemical test to assess postharvest deterioration of sugarcane and milled juice[J].Sugar Tech, 2009, 11(2): 189-195. DOI:10.1007/s12355-009-0030-8.
[10] BHATIA S, BHAKRI G, ARORA M, et al. Kinetic and thermodynamic properties of partially purified dextranase from Paecilomyces lilacinus,and its application in dextran removal from cane juice[J]. Sugar Tech,2015, 18(2): 204-213. DOI:10.1007/s12355-015-0378-x.
[11] MAZZAOUI S A, BURROW M F, TYAS M J, et al. Incorporation of casein phosphopeptide-amorphous calcium phosphate into a glass-ionomer cement[J]. Dental Materials, 2011, 27(3): 235-243.DOI:10.1177/154405910308201113.
[12] 李卫娟, 焦豫良, 吕明生, 等. 一株产右旋糖苷酶海洋细菌的筛选鉴定[J]. 微生物学通报, 2013, 40(5): 756-765.
[13] 张宇琪. 右旋糖酐酶产生菌的筛选、酶学性质及其应用研究[D].合肥: 合肥工业大学, 2016.
[14] WANG D, LU M, WANG S, et al. Purification and characterization of a novel marine Arthrobacter oxydans KQ11 dextranase[J]. Carbohydrate Polymers, 2014, 106(1): 71. DOI:10.1016/j.carbpol.2014.01.102.
[15] WANG D, LU M, WANG X, et al. Improving stability of a novel dextran-degrading enzyme from marine Arthrobacter oxydans KQ11[J]. Carbohydrate Polymers, 2014, 103(1): 294-299.DOI:10.1016/j.carbpol.2013.12.025.
[16] MOON Y H, MADSEN L, CHUNG C H, et al. Lime application for the efficient production of nutraceutical glucooligosaccharides from Leuconostoc mesenteroides, NRRL B-742 (ATCC13146)[J]. Journal of Industrial Microbiology & Biotechnology, 2015, 42(2): 279-285.DOI:10.1007/s10295-014-1568-3.
[17] REN W, WANG S, LÜ M, et al. Optimization of four types of antimicrobial agents to increase the inhibitory ability of marine Arthrobacter oxydans KQ11 dextranase mouthwash[J]. Chinese Journal of Oceanology and Limnology, 2016, 34(2): 354-366.
[18] LARSSON A M, ANDERSSON R, STÅHLBERG J, et al. Dextranase from Penicillium minioluteum: reaction course, crystal structure, and product complex[J]. Structure, 2003, 11(9): 1111-1121. DOI:10.1016/S0969-2126(03)00147-3.
[19] SUZUKI N, KIM Y M, FUJIMOTO Z, et al. Structural elucidation of dextran degradation mechanism by Streptococcus mutans dextranase belonging to glycoside hydrolase family 66[J]. Journal of Biological Chemistry, 2012, 287(24): 19916. DOI:10.1074/jbc.M112.342444.
[20] SUZUKI N, KISHINE N, FUJIMOTO Z, et al. Crystal structure of thermophilic dextranase from Thermoanaerobacter pseudethanolicus[J]. Journal of Biochemistry, 2015, 159(3): 331-339.DOI:10.1093/jb/mvv104.
[21] OKAZAWA Y, MIYAZAKI T, YOKOI G, et al. Crystal structure and mutational analysis of isomaltodextranase, a member of glycoside hydrolase family 27[J]. Journal of Biological Chemistry, 2015,290(43): 26339-26349. DOI:10.1074/jbc.M115.680942.
[22] MIZUNO M, KOLDE A, YAMAMURA A, et al. Crystal structure of Aspergillus niger isopullulanase, a member of glycoside hydrolase family 49[J]. Journal of Molecular Biology, 2008, 376(1): 210-220.DOI:10.1016/j.jmb.2007.11.098.
[23] 黄红卫, 刘艳丽, 李春. 糖苷酶的研究及其改造策略[J]. 生物技术通报, 2010(5): 55-60.
[24] YANG Q, WANG B C, ZHANG Z, et al. The effects of macromolecular crowding and surface charge on the properties of an immobilized enzyme: activity, thermal stability, catalytic efficiency and reusability[J]. RSC Advances, 2017, 7(60): 38028-38036.DOI:10.1039/C7RA06544B.
[25] WALL M E. Internal protein motions in molecular-dynamics simulations of bragg and diffuse X-ray scattering[J]. 2018, 5(2):172-181. DOI:10.1107/S2052252518000519.
[26] AKEBOSHI H, TONOZUKA T, FURUKAWA T, et al. Insights into the reaction mechanism of glycosyl hydrolase family 49[J]. European Journal of Biochemistry, 2004, 271(22): 4420-4427. DOI:10.1111/j.1432-1033.2004.04378.x.
[27] SU H, SHENG X, LIU Y. Insights into the catalytic mechanism of N-acetylglucosaminidase glycoside hydrolase from Bacillus subtilis: a QM/MM study[J]. Organic & Biomolecular Chemistry, 2016, 14(13):3432. DOI:10.1039/C6OB00320F.
[28] BAI Y, GANGOITI J, DIJKSTRA B W, et al. Crystal structure of 4,6-α-glucanotransferase supports diet-driven evolution of GH70 enzymes from α-amylases in oral bacteria[J]. Structure, 2016, 25(2):231-242. DOI:10.1016/j.str.2016.11.023.
[29] MHLONGO N N, SKELTON A A, KRUGER G, et al. A critical survey of average distances between catalytic carboxyl groups in glycoside hydrolases[J]. Proteins-Structure Function & Bioinformatics,2014, 82(9): 1747-1755. DOI:10.1002/prot.24528.
[30] RIGDEN D J, FRANCO O L. Beta-helical catalytic domains in glycoside hydrolase families 49, 55 and 87: domain architecture,modelling and assignment of catalytic residues[J]. FEBS Letters, 2002,530(1/2/3): 225-232. DOI:10.1016/S0014-5793(02)03490-7.
Dextranase from Arthrobacter oxydans KQ11: Identification of Key Residues in Catalysis
LIU Le1, DING Yi1, WANG Zixuan1, FANG Yaowei1, WANG Shujun1,2, LÜ Mingsheng1,*
(1. College of Marine Life and Fisheries, Huaihai Institute of Technology, Lianyungang 222005, China;2. Marine Resources Development Institute of Jiangsu, Lianyungang 222005, China)
Abstract: The catalytic domain and key amino acid residues of dextranase from Arthrobacter oxydans KQ11 (AoDex) were predicted by homologous sequence alignment. The mutants Gln418→Gly, Asp420→Gly, Glu423→Gly, Asp439→Gly, and Asp440→Gly were obtained from the catalytic domain 418-QTDGIELYKGSTMKNTFFNANDD-440 by site-directed mutagenesis. The mutant dextranases Q418GDex, D420GDex, E423GDex, and D439GDex had almost no enzymatic activity, while D440GDex had equal enzymatic activity to AoDex. However, at temperatures between 25 and 40 ℃,D440GDex activity increased by 2-3 folds compared to AoDex activity. The optimum pH for D440GDex was 6.5 while that for AoDex was 5.5. Q418, D420, E423, D439 were the key amino acid residues in the catalytic domain of AoDex. Mutation of D440 had a great impact on the enzymatic properties, suggesting it to be not the general base of the AoDex catalytic domain. These fi ndings suggest that AoDex and the GH family 49 share a similar catalytic mechanism, which will provide theoretical support for improving the enzymatic properties of AoDex.
Keywords: dextranase; site-directed mutagenesis; catalytic domain; key amino acid residues; structure prediction
收稿日期:2018-03-05
基金项目:国家自然科学基金面上项目(31471719);江苏省科技厅社会发展项目(BE2016702)
第一作者简介:刘乐(1991—)(ORCID: 0000-0001-9572-3057),男,硕士研究生,研究方向为酶的性质与应用。E-mail: liulezz3@163.com
*通信作者简介:吕明生(1963—)(ORCID: 0000-0003-4409-3766),男,教授,博士,研究方向为酶的性质与应用、纳米材料。E-mail: mingshenglu@hotmail.com
DOI:10.7506/spkx1002-6630-20180305-031
中图分类号:Q812
文献标志码:A
文章编号:1002-6630(2019)06-0113-08
引文格式:刘乐, 丁一, 王紫玄, 等. 海洋氧化节杆菌KQ11右旋糖苷酶催化位点关键氨基酸[J]. 食品科学, 2019, 40(6): 113-120.DOI:10.7506/spkx1002-6630-20180305-031. http://www.spkx.net.cn
LIU Le, DING Yi, WANG Zixuan, et al. Dextranase from Arthrobacter oxydans KQ11: identification of key residues in catalysis[J]. Food Science, 2019, 40(6): 113-120. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-20180305-031. http://www.spkx.net.cn