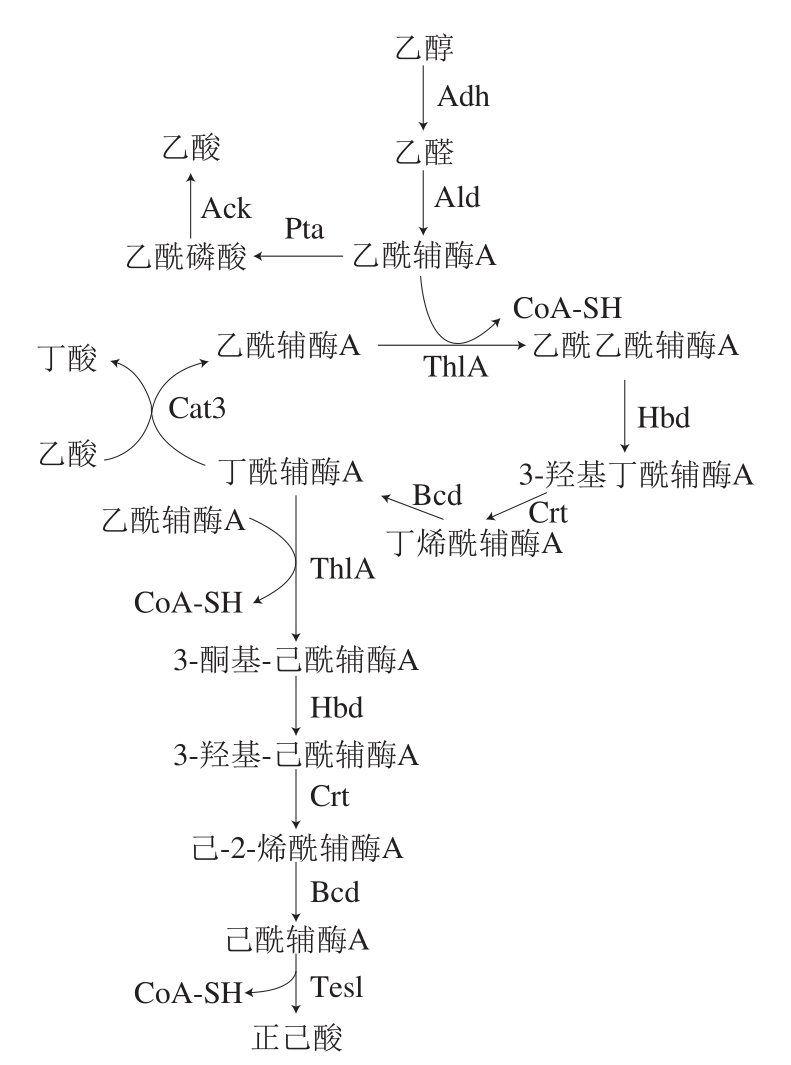
图1 C. kluyveri的己酸代谢途径
Fig. 1 Hexanoic acid metabolic pathway in C. kluyveri
Adh.乙醇脱氢酶;Ald.乙醛脱氢酶;Pta.磷酸转乙酰酶;Ack.乙酸激酶;ThlA.乙酰乙酰辅酶A乙酰转移酶(硫解酶);Hbd. 3-羟基丁酰辅酶A脱氢酶;Crt. 3-羟基丁酰辅酶A脱水酶;Bcd.丁酰辅酶A脱氢酶;Cat3.丁酰辅酶A:乙酸辅酶A转移酶;Tes1.酰基辅酶A硫酯酶。
白酒是中国特色发酵酒类饮品,也是世界知名蒸馏酒之一[1]。依据香气特征,中国白酒分为12 种香型,其中浓香型白酒目前销量占白酒市场份额的70%以上,属于白酒行业的支柱型产品[1-2]。研究发现,浓香型白酒的主体风味物质为己酸乙酯。国家标准规定,优级高度酒的总酯质量浓度不低于2.0 g/L,其中己酸乙酯质量浓度不低于1.2 g/L,优级低度酒的总酯质量浓度不低于1.5 g/L,其中己酸乙酯质量浓度不低于0.7 g/L[3]。生产实践中,以己酸乙酯含量的高低作为浓香型白酒品质的重要衡量参数已成为业内共识[4-6]。这充分说明己酸乙酯对浓香型白酒品质的重要作用。己酸乙酯合成的前体物质为己酸和乙醇。其中,白酒酿造过程己酸的合成代谢对于己酸乙酯的生成具有决定性的作用[7]。研究发现,浓香型白酒酿造过程Clostridium属的微生物具有较高丰度,且对于酒醅己酸的产生具有重要贡献[7]。其中,Clostridium kluyveri是白酒来源Clostridium属微生物的典型代表[8]。20世纪60年代,C. kluyveri自白酒窖泥被分离,并经研究证实对于己酸代谢合成和己酸乙酯生成具有显著促进作用,继而通过微生物的外源添加,保障白酒酿造过程己酸乙酯的生成,使酒体呈现浓香型白酒的感官和风味,进而推动浓香型白酒的生产突破传统白酒酿造的地域依赖性,目前全国多个地区均能够生产优质浓香型白酒[1]。这也充分说明微生物对于白酒酿造的重要性。
C. kluyveri是己酸的重要代谢生产菌种之一,所代谢合成的己酸具有重要用途。作为六碳的小分子脂肪酸,己酸可用于香料、医药前体、食品添加剂、润滑油、烟草调香、橡胶和印染领域,同时可作为前体合成己酸酯如己酸乙酯,是食品领域的重要风味酯类,用途广泛[9-12]。因此,有研究对微生物的己酸代谢进行探索,如通过基因组测序和注释,解析己酸代谢途径等[8,10]。另有文献报道,Clostridium属的其他菌株、Megasphaera elsdenii、基因工程改造的Escherichia coli和Kluyveromyces marxianus等也具有己酸的代谢合成能力[9-12]。但目前研究显示,C. kluyveri己酸代谢合成的产量和生产速率均居于前列[10]。由于己酸广泛的用途和C.kluyveri突出的己酸代谢合成性能,C. kluyveri的己酸代谢已有文献报道。Seedorf等[8]解析C. kluyveri模式菌株DSM 555的基因组信息,对乙醇代谢合成丁酸的途径进行详细分析。之后,Tao Yong等[10]对另一株己酸高产菌株Clostridium sp. CPB6的基因组序列进行解析和注释,并分析该菌株代谢合成己酸的可能途径。
目前研究认为,C. kluyveri合成己酸的经典代谢过程是通过乙醇作为底物和电子供体的反向β-氧化循环链延长机制实现的。其代谢途径见图1。乙醇首先经乙醇脱氢酶(Adh)转化为乙醛,经乙醛脱氢酶(Ald)转化为乙酰辅酶A,这两步转化均偶合NAD+转化为NADH。乙酰辅酶A经磷酸转乙酰酶(Pta)生成乙酰磷酸,经乙酸激酶(Ack)转化生成乙酸,该步催化伴随ADP生成ATP。乙酰辅酶A还可经乙酰辅酶A乙酰转移酶(又名硫解酶)(ThlA)转化为乙酰乙酰辅酶A,经3-羟基丁酰辅酶A脱氢酶(Hbd)转化为3-羟基丁酰辅酶A并消耗NADH。3-羟基丁酰辅酶A经3-羟基丁酰辅酶A脱水酶(Crt)转化为丁烯酰辅酶A。丁烯酰辅酶A在丁酰辅酶A脱氢酶(Bcd)的催化下,转化为丁酰辅酶A,该步催化偶合FAD依赖型的电子转移黄素蛋白(Etf)和铁氧化还原蛋白氧化还原酶(Rnf),并伴随电子的转移。丁酰辅酶A和乙酸经丁酰辅酶A:乙酸辅酶A转移酶(Cat3)转化为乙酰辅酶A和丁酸,实现以乙酸合成丁酸的过程。再经类似的催化过程,实现由丁酰辅酶A到己酰辅酶A,经酰基辅酶A硫酯酶(Tes1)催化生成己酸。代谢途径分析表明,由乙酰辅酶A到己酸代谢的主要催化酶包括乙酰辅酶A乙酰转移酶/硫解酶(ThlA)、3-羟基丁酰辅酶A脱氢酶(Hbd)、3-羟基丁酰辅酶A脱水酶(Crt)和丁酰辅酶A脱氢酶(Bcd)。
基于代谢途径的解析,利用代谢工程技术进行微生物的遗传改造,改良微生物的代谢特性,是实现目标化合物微生物高产的有效途径。然而,由于C. kluyveri遗传操作体系的匮乏,目前通过基因工程手段利用微生物进行己酸的生产优化主要集中于在大肠杆菌或者酵母菌株中异源重构己酸的代谢途径来实现[11-12]。相关研究发现,乙酰辅酶A乙酰转移酶是己酸代谢途径的关键酶,其表达强度对于己酸代谢的产量和速率具有决定作用[12]。然而到目前为止,已知的C. kluyveri基因组测序和注释结果关于己酸代谢核心催化酶以及关键酶的注释及功能分析仍不深入,甚至存在欠缺和谬误,不利于后续对己酸代谢的深入研究。基于此,本研究通过比较基因组学和生物信息学相结合的方法,对3 株遗传信息全面解析的C. kluyveri进行己酸代谢途径核心催化酶的重新注释和关键酶硫解酶的结构及催化机制分析,为后续的深入研究提供参考。
图1 C. kluyveri的己酸代谢途径
Fig. 1 Hexanoic acid metabolic pathway in C. kluyveri
Adh.乙醇脱氢酶;Ald.乙醛脱氢酶;Pta.磷酸转乙酰酶;Ack.乙酸激酶;ThlA.乙酰乙酰辅酶A乙酰转移酶(硫解酶);Hbd. 3-羟基丁酰辅酶A脱氢酶;Crt. 3-羟基丁酰辅酶A脱水酶;Bcd.丁酰辅酶A脱氢酶;Cat3.丁酰辅酶A:乙酸辅酶A转移酶;Tes1.酰基辅酶A硫酯酶。
检索NCBI的基因组数据库,共获得已知全基因组信息的3 株C. kluyveri,编号分别为DSM 555[8]、NBRC 12016[13]和JZZ[12],以其为研究对象,将全基因组序列信息下载到本地进行分析。
1.2.1 比较基因组学分析
利用软件MAUVE 2.3.1进行3 株C. kluyveri菌株全基因组序列分析[14]。利用文本编辑软件fasta编辑符合比对要求的基因组序列文件,通过MAUVE细菌基因组比对默认参数进行比较基因组学分析。
1.2.2 关键酶序列和基本性质分析
序列比对选用DNAMAN 7.0软件完成、酶的基本性质分析采用ExPASy-ProtParam tool完成(http://web.expasy.org/protparam/)[15]。
蛋白质系统发育树构建:选取代表性的不同乙酰辅酶A乙酰转移酶序列,按照系统发育分析软件MEGA 5.0的要求制作序列比对文件,通过Neighbour-Joining算法构建系统发育树,Bootstrap values:1 000[16]。
1.2.3 关键酶亲水/疏水性分析、信号肽和跨膜区预测
蛋白亲水/疏水性分析通过软件ExPASy-ProtScale完成(http://web.expasy.org/protscale/)[15],信号肽的预测通过SignalP 4.1网站进行(http://www.cbs.dtu.dk/services/SignalP/)[17],利用在线软件TMHMM 2.0进行蛋白跨膜区的预测(http://www.cbs.dtu.dk/services/TMHMM)。
1.2.4 关键酶二级、三维结构预测和催化机制分析
蛋白二级结构利用软件SOPMA(Secondary Structure Prediction Method)(https://npsa-prabi.ibcp.fr/ cgi-bin/npsa_automat.pl?page=npsa_sopma.html)进行分析[18]。蛋白三维结构预测通过SWISS-MODEL(http://swissmodel.expasy.org/)完成[19]。分子对接分析酶与底物作用关系通过DISCOVERY STUDIO 2.5软件完成[20]。
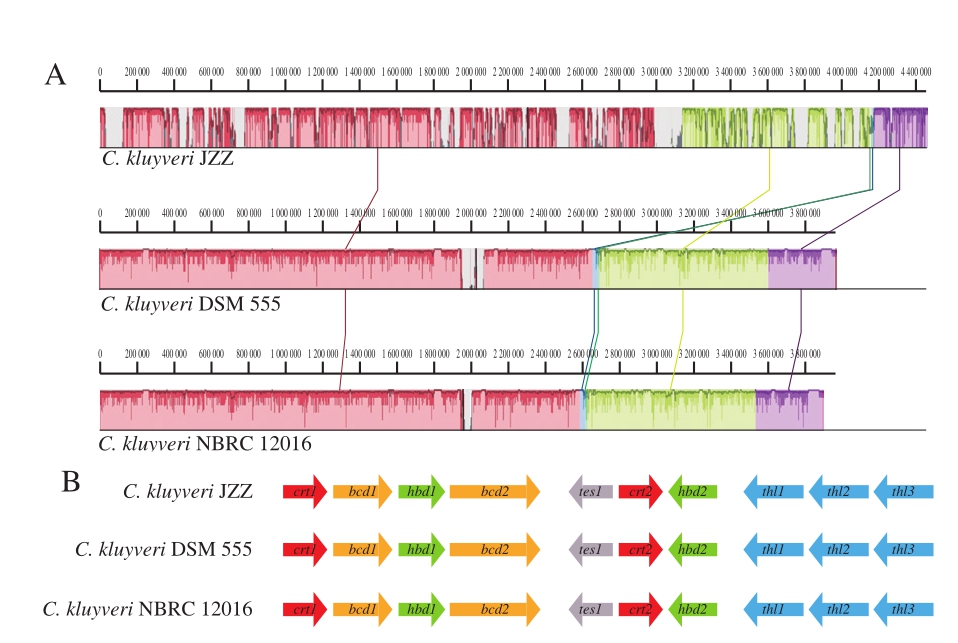
图2 3 株C. kluyveri全基因组序列比对(A)和己酸代谢途径基因分布分析(B)
Fig. 2 Whole genome sequence alignment (A) of three C. kluyveri and gene distribution of hexanoic acid metabolic pathway
比较基因组分析发现,模式菌株DSM 555与NBRC 12016的基因组长度接近,但均明显短于JZZ,说明DSM 555与NBRC 12016在进化关系上更加接近,而JZZ可能存在区别于DSM 555和NBRC 12016的独有遗传信息(图2A)。文献研究表明,菌株JZZ分离自白酒酿造过程,该菌株基因组独有的遗传信息可能与基因水平转移有关[21],来自白酒酿造过程中其他微生物所携带的遗传信息,赋予该菌株区别于DSM 555和NBRC 12016的生物学功能,使其在白酒酿造的独特生境下可以更好地生存和繁殖。另外,比较基因组学分析表明,3 株菌株均不存在明显的基因组DNA片段的转换或者颠换。基于比较基因组学分析和基因组基因注释结果,研究绘制不同拷贝的己酸代谢核心催化酶编码基因在基因组上的相对位置图谱(图2B)。3 株菌株均携带己酸代谢的核心催化酶编码基因,表明具有代谢合成己酸的能力,且每一株菌对应核心催化酶编码基因的拷贝数一致,但同一株菌不同酶编码基因的拷贝数不同(图2B)。3 株菌株携带己酸代谢的核心催化酶编码基因均为多拷贝,说明该代谢途径属于C. kluyveri的关键代谢途径,对于维持菌株的生存和基本代谢至关重要。
表1 3 株C. kluyveri己酸代谢途径的核心催化酶重新注释
Table 1 Re-annotation of the core enzymes of the hexanoic acid metabolic pathway in threeC. kluyveri strains
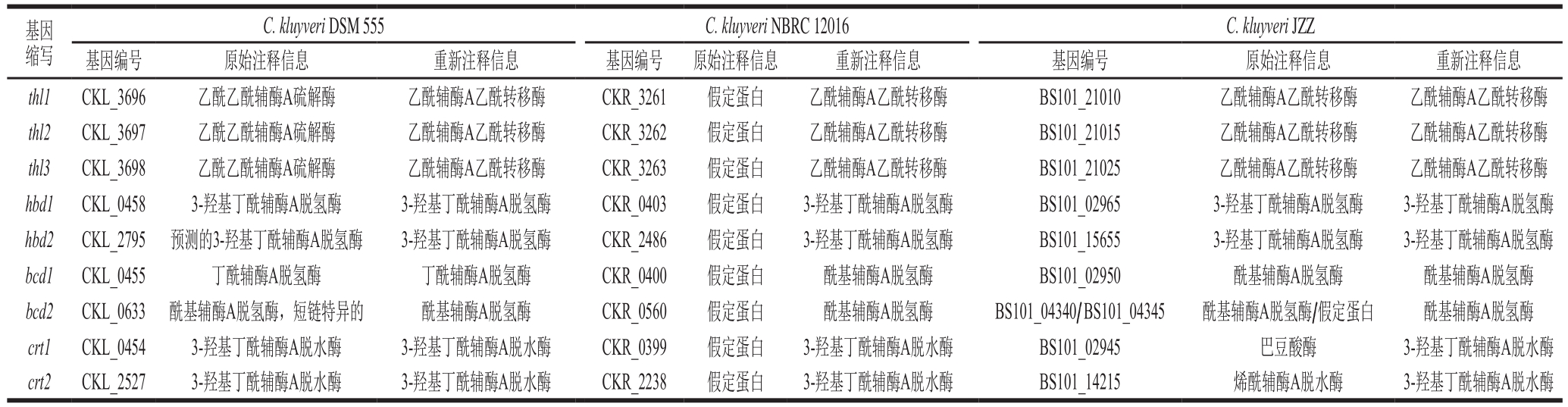
基因缩写C. kluyveri JZZ基因编号 原始注释信息 重新注释信息 基因编号 原始注释信息 重新注释信息 基因编号 原始注释信息 重新注释信息thl1 CKL_3696 乙酰乙酰辅酶A硫解酶 乙酰辅酶A乙酰转移酶 CKR_3261 假定蛋白 乙酰辅酶A乙酰转移酶 BS101_21010 乙酰辅酶A乙酰转移酶 乙酰辅酶A乙酰转移酶thl2 CKL_3697 乙酰乙酰辅酶A硫解酶 乙酰辅酶A乙酰转移酶 CKR_3262 假定蛋白 乙酰辅酶A乙酰转移酶 BS101_21015 乙酰辅酶A乙酰转移酶 乙酰辅酶A乙酰转移酶thl3 CKL_3698 乙酰乙酰辅酶A硫解酶 乙酰辅酶A乙酰转移酶 CKR_3263 假定蛋白 乙酰辅酶A乙酰转移酶 BS101_21025 乙酰辅酶A乙酰转移酶 乙酰辅酶A乙酰转移酶hbd1 CKL_0458 3-羟基丁酰辅酶A脱氢酶 3-羟基丁酰辅酶A脱氢酶 CKR_0403 假定蛋白 3-羟基丁酰辅酶A脱氢酶 BS101_02965 3-羟基丁酰辅酶A脱氢酶 3-羟基丁酰辅酶A脱氢酶hbd2 CKL_2795 预测的3-羟基丁酰辅酶A脱氢酶 3-羟基丁酰辅酶A脱氢酶 CKR_2486 假定蛋白 3-羟基丁酰辅酶A脱氢酶 BS101_15655 3-羟基丁酰辅酶A脱氢酶 3-羟基丁酰辅酶A脱氢酶bcd1 CKL_0455 丁酰辅酶A脱氢酶 丁酰辅酶A脱氢酶 CKR_0400 假定蛋白 酰基辅酶A脱氢酶 BS101_02950 酰基辅酶A脱氢酶 酰基辅酶A脱氢酶bcd2 CKL_0633 酰基辅酶A脱氢酶,短链特异的 酰基辅酶A脱氢酶 CKR_0560 假定蛋白 酰基辅酶A脱氢酶 BS101_04340/ BS101_04345 酰基辅酶A脱氢酶/假定蛋白 酰基辅酶A脱氢酶crt1 CKL_0454 3-羟基丁酰辅酶A脱水酶 3-羟基丁酰辅酶A脱水酶 CKR_0399 假定蛋白 3-羟基丁酰辅酶A脱水酶 BS101_02945 巴豆酸酶 3-羟基丁酰辅酶A脱水酶crt2 CKL_2527 3-羟基丁酰辅酶A脱水酶 3-羟基丁酰辅酶A脱水酶 CKR_2238 假定蛋白 3-羟基丁酰辅酶A脱水酶 BS101_14215 烯酰辅酶A脱水酶 3-羟基丁酰辅酶A脱水酶C. kluyveri DSM 555C. kluyveri NBRC 12016
表2 关键硫解酶基本性质分析
Table 2 Characteristics of the key thiolases
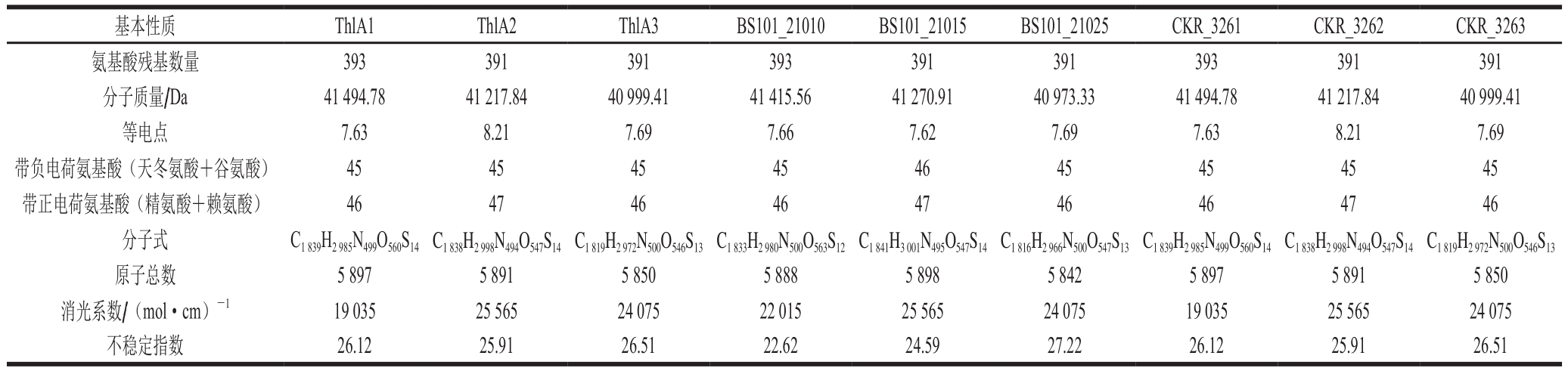
基本性质 ThlA1 ThlA2 ThlA3 BS101_21010 BS101_21015 BS101_21025 CKR_3261 CKR_3262 CKR_3263氨基酸残基数量 393 391 391 393 391 391 393 391 391分子质量/Da 41 494.78 41 217.84 40 999.41 41 415.56 41 270.91 40 973.33 41 494.78 41 217.84 40 999.41等电点 7.63 8.21 7.69 7.66 7.62 7.69 7.63 8.21 7.69带负电荷氨基酸(天冬氨酸+谷氨酸) 45 45 45 45 46 45 45 45 45带正电荷氨基酸(精氨酸+赖氨酸) 46 47 46 46 47 46 46 47 46分子式 C1 839H2 985N499O560S14 C1 838H2 998N494O547S14 C1 819H2 972N500O546S13 C1 833H2 980N500O563S12 C1 841H3 001N495O547S14 C1 816H2 966N500O547S13 C1 839H2 985N499O560S14 C1 838H2 998N494O547S14 C1 819H2 972N500O546S13原子总数 5 897 5 891 5 850 5 888 5 898 5 842 5 897 5 891 5 850消光系数/(mol·cm)-1 19 035 25 565 24 075 22 015 25 565 24 075 19 035 25 565 24 075不稳定指数 26.12 25.91 26.51 22.62 24.59 27.22 26.12 25.91 26.51
对核心代谢酶的注释信息进行比较分析,由表1可知,NBRC 12016由于基因组测序较早,非冗余蛋白质数据库的信息相对不完善,导致己酸代谢途径的注释信息匮乏。与之相比,DSM 555和JZZ的注释信息较为完善,但JZZ存在酰基辅酶A脱氢酶注释的错误,编码酰基辅酶A脱氢酶的基因被错误预测为编号BS101_04340和BS101_04345的两个基因,对应编码蛋白的注释信息亦需更正。另外,基因bcd2编码蛋白的注释信息为酰基辅酶A脱氢酶、短链特异的,表明源于DSM 555的基因bcd1和bcd2编码蛋白的底物特异性可能存在差别,有待后续研究证实。由于己酸代谢合成是脂肪酸代谢链延长的过程,C. kluyveri携带的己酸代谢关键酶硫解酶的序列多态性可能与催化过程链延长循环的底物特异性有关,但目前鲜见相关的研究报道。
对3 株C.kluyveri菌株己酸代谢途径的关键酶乙酰辅酶A乙酰转移酶(硫解酶)进行基本酶学性质分析,包括氨基酸残基数量、分子质量、等电点、带负电荷氨基酸、带正电荷氨基酸、分子式、原子总数、消光系数和不稳定指数(表2)。不同菌株来源对应的硫解酶以及同一菌株来源不同拷贝的硫解酶的基本性质包括分子质量、等电点、分子式、原子总数、消光系数和不稳定指数均存在区别。其中,不同硫解酶的不稳定指数均小于40,说明蛋白本身在结构上是稳定的[22]。蛋白的稳定性与序列的二肽组成紧密相关[22],不同硫解酶的序列存在差别(图3),导致氨基酸序列中的二肽组成具有差异,是稳定性差别的内在原因。另外,ThlA1、ThlA3、BS101_21010、BS101_21025、CKR_3261和CKR_3263虽然带正负电荷氨基酸总数分别相等,但是经统计,组成正负电荷的氨基酸不同(图3),不同氨基酸的解离系数存在差别,导致酶的等电点存在微小差别。上述分析对于后续该类关键酶的分离纯化和催化特性研究具有一定的指导意义。
在硫解酶基本理化性质解析基础上,研究对酶的氨基酸序列进行比对和系统发育分析。结果表明,C. kluyveri不同种均携带3 个拷贝的硫解酶,同一菌株来源的3 个硫解酶的序列和不同菌株来源对应硫解酶的序列均存在差别(图3),且不同来源硫解酶在系统发育树上明显聚于不同的分支(图4)。同时,C. kluyveri来源硫解酶与Clostridium属其余种来源的硫解酶具有序列的差异。硫解酶催化短链的酰基辅酶A合成长链的酰基辅酶A,是脂肪酸合成代谢的关键酶之一[23]。研究发现,微生物中脂肪酸合成代谢一般存在3 种类型的硫解酶,I型脂肪酸合成酶(fatty-acid synthesis type I,FAS I),II型脂肪酸合成酶(FAS II)和III型脂肪酸合成酶(FAS III)。其中,FAS I和FAS II与脂肪酸的链延长相关,而FAS III与脂肪酸合成的反应起始相关[23-24]。由于C. kluyveri来源硫解酶的研究较少,目前尚鲜见关于不同拷贝硫解酶底物特异性的文献报道。图4表明,以C. kluyveri来源硫解酶为对象进行深入研究,对于解析C. kluyveri己酸代谢途径具有重要意义。

图3 关键硫解酶序列比对
Fig. 3 Sequence alignment of the key thiolases
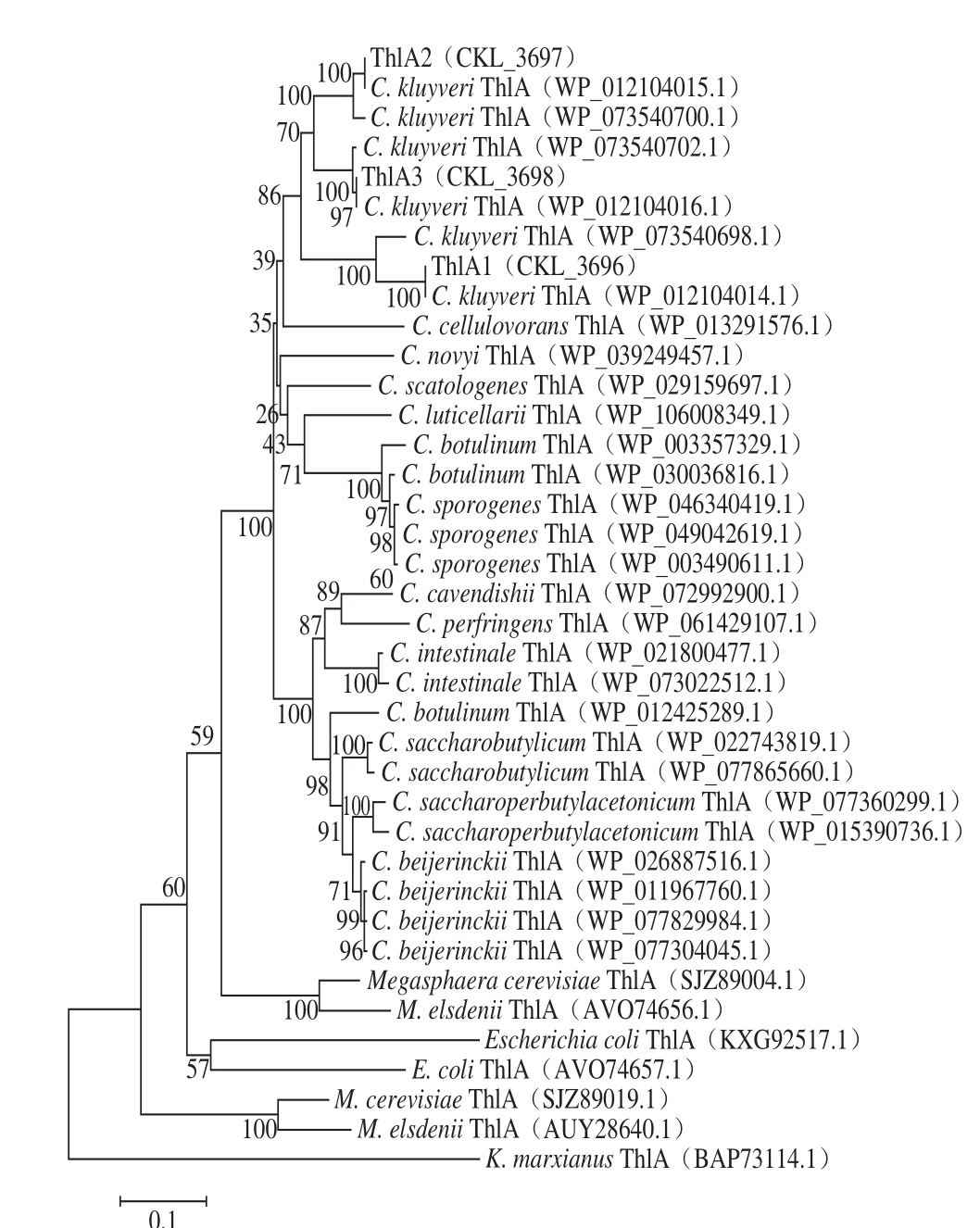
图4 关键硫解酶系统发育分析
Fig. 4 Phylogenetic analysis of the key thiolases
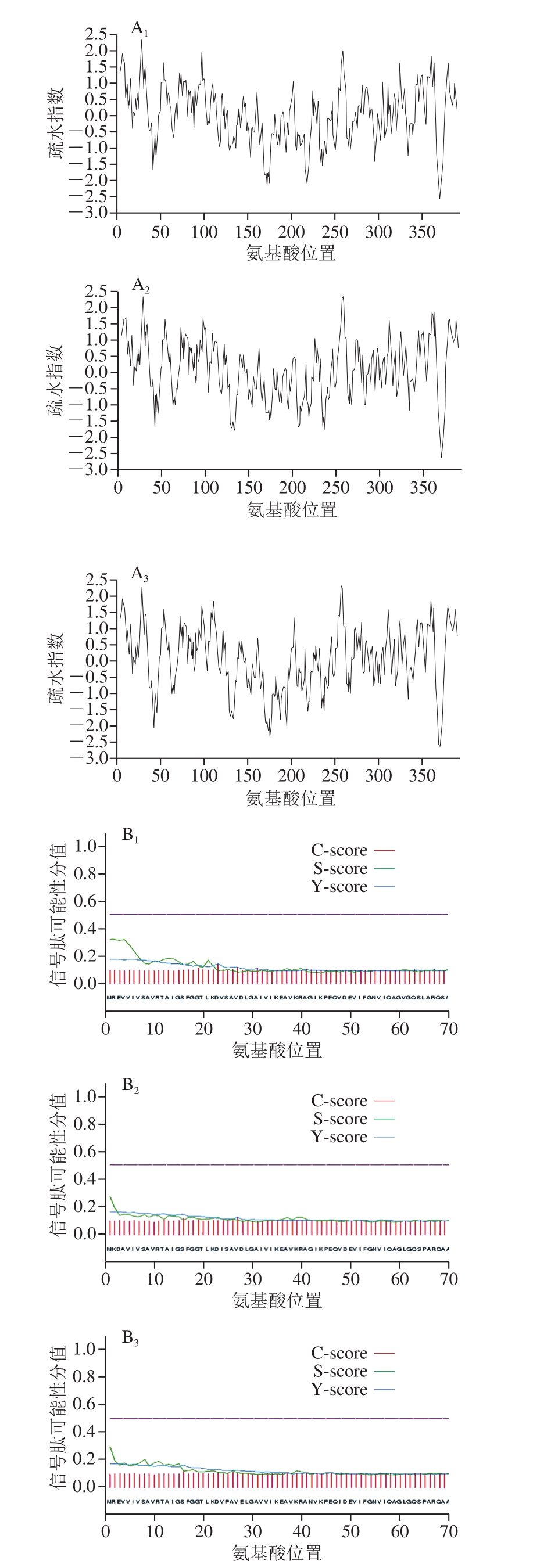
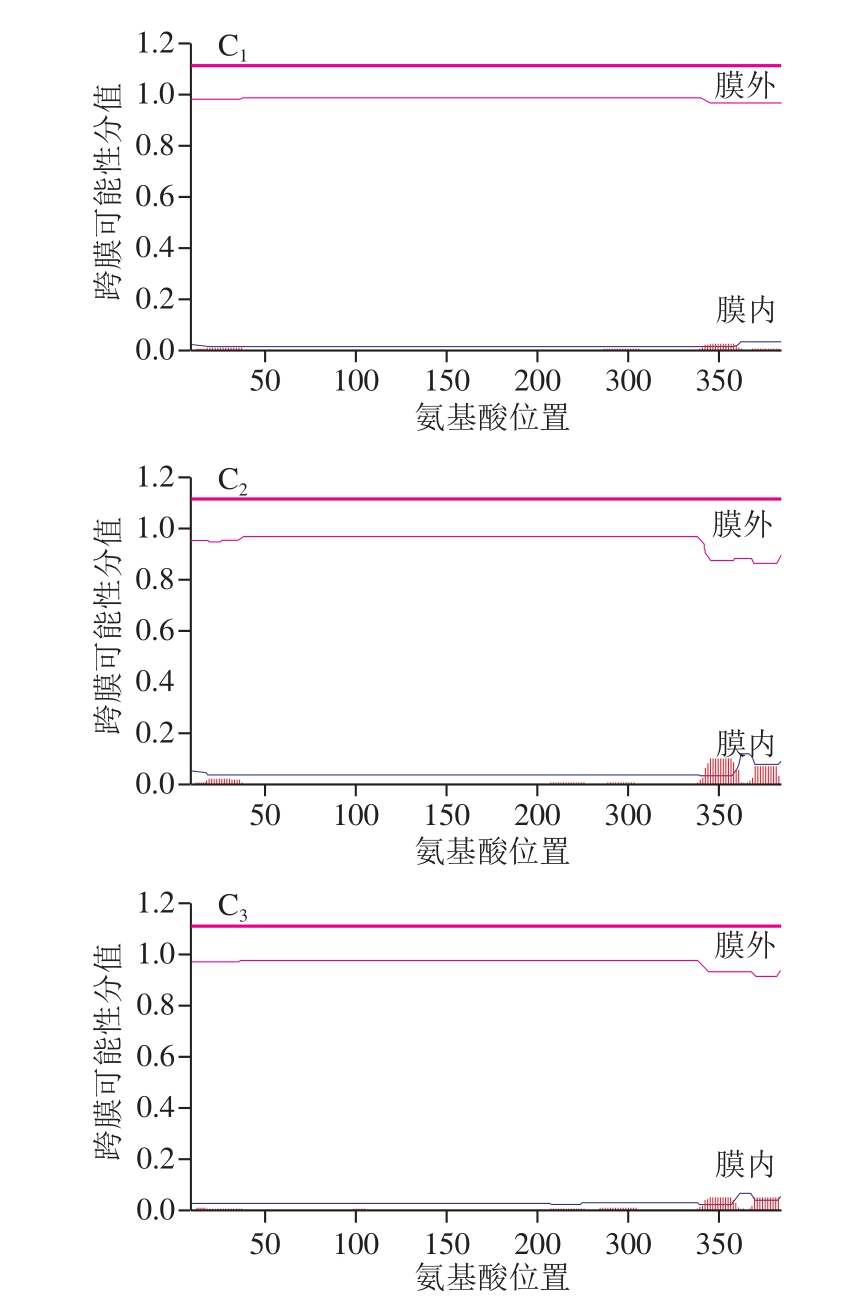
图5 C. kluyveri DSM 555关键酶ThlA疏水性分析(A)、信号肽(B)和跨膜区(C)预测
Fig. 5 Hydrophobic analysis (A), signal peptide (B) and transmembrane domain (C) prediction of the key enzymes ThlA from C. kluyveri DSM 555
A1~A3. ThlA1、ThlA2、ThlA3疏水性。B1~B3. ThlA1、ThlA2、ThlA3信号肽预测;C-score.预测的切割位点值;S-score.预测的信号肽值;Y-score. C-score与S-score平滑导数之间的几何平均值。C1~C3. ThlA1、ThlA2、ThlA3跨膜区预测。
在酶的理化性质和系统发育分析基础上,研究以C.kluyveri模式菌株DSM 555来源硫解酶ThlA为代表性对象,采用Hphob/Kyte & Doolittle算法计算ThlA1、ThlA2和ThlA3的疏水性(图5A)[25]。ThlA1疏水指数最小值为-2.556(370位),最大值为2.322(30位),总平均亲水性指数(grand average of hydropathicity,GRAVY)为0.049;ThlA2疏水指数最小值为-2.622(368位),最大值为2.322(30位),GRAVY为0.104;ThlA3疏水指数最小值为-2.622(368位),最大值为2.289(30位),GRAVY为0.076。特定位点的氨基酸疏水性指数越大,表明该位点的疏水性越高[25]。3 个ThlA的GRAVY存在差别,说明三者虽然都是疏水蛋白,但是疏水性有差别。蛋白质的疏水作用与其稳定性具有密切的相关性,通过蛋白氨基酸位点的疏水性分析,结合蛋白质工程技术,进行关键位点或者区域的理性设计和改造,对于改善蛋白的稳定性具有重要作用[26]。图5B表明,3 拷贝的ThlA均不具有信号肽,不能分泌到胞外,是具有重要催化功能的胞内酶。跨膜区预测显示,3 个酶均不具有明显的跨膜区,是非跨膜蛋白(图5C)。但与ThlA1相比,ThlA2和ThlA3的C端具有更强的疏水性,疏水性的差别可能会影响蛋白的结构,进而影响酶的催化特性[27]。不同硫解酶的催化特性存在差异,最近研究发现可能与硫解酶C端loop区的空间结构有关[28],但C端疏水性对催化的具体影响仍需要通过实验加以解析。通过生物信息学的技术手段对酶的可能催化机制进行先期分析,可为后续的研究提供借鉴。
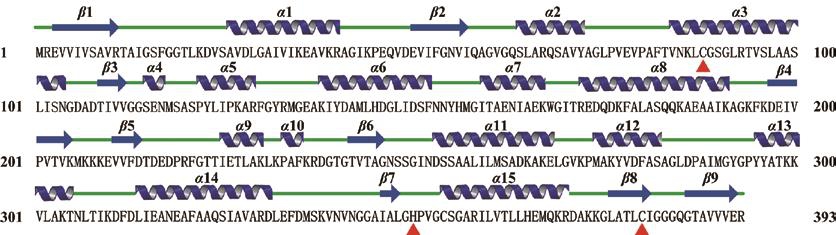
图6 关键酶ThlA1二级结构预测
Fig. 6 Prediction of the secondary structure of the key enzyme ThlA1
以C. kluyveri DSM 555来源ThlA1为例,进行酶的结构和催化机制解析。二级结构预测表明,ThlA1蛋白具有15 个α-螺旋和9 个β-折叠,占序列总长的60.3%。另经与相近序列的比对分析,确定Cys88、His350和Cys380为催化关键位点(图6三角标示位点的氨基酸),其中,His350行使激活酶催化的功能;Cys88与Cys380可以形成二硫键,二硫键的形成或者断开会直接影响酶的催化,是调节酶催化与否的关键位点[23,28]。研究发现,绿色下划线部分的loop结构在空间上位于Cys88和Cys380之间,当二硫键形成时,loop区会发生空间上的旋转,进一步阻碍乙酰辅酶A与酶催化活性中心的结合[28]。二级结构的预测对于蛋白的研究具有重要帮助,例如,当序列同源性较低时,二级结构的预测有助于确定蛋白质之间结构与功能的关系;借助于蛋白二级结构统计分析的规律,可以用于全新蛋白的从头设计;另外,二级结构的预测有助于多维核磁共振中二级结构的确认和晶体结构的解析[26,29]。
利用软件SWISS MODEL以C. acetobutylicum来源硫解酶CaTHL(PDB:4xl4.1.A)为模板,构建三维结构模型(图7A)。4xl4.1.A与ThlA1序列相似度为72.82%。目前研究发现,硫解酶一般以四聚体的形式存在,利用SWISS MODEL同时进行了四聚体结构的预测(参考蛋白PDB:4wyr.1.A)(图7D)。四聚体是通过单体的L-结构域(图7A两β折叠及其之间的无规则卷曲)结合形成的规则对称结构。研究同时进行分子对接,确定酶与底物分子乙酰辅酶A的最佳结合角度和取向,并分析酶与底物可能的成键(图7B和7C)。结构解析结合文献总结表明,Clostridium属微生物来源的ThlA具有不同于已知硫解酶的独特调控催化机制——氧化还原开关调控机制[28]。当酶处于还原状态时,Cys88与Cys380保持还原状态,之间不形成二硫键,Cys88的硫原子失去氢离子被激活,进而起始催化反应(图7E I);当酶处于氧化状态时,Cys88与Cys380失去氢离子并形成二硫键,酶处于无活性状态,不行使催化反应(图7E II)。
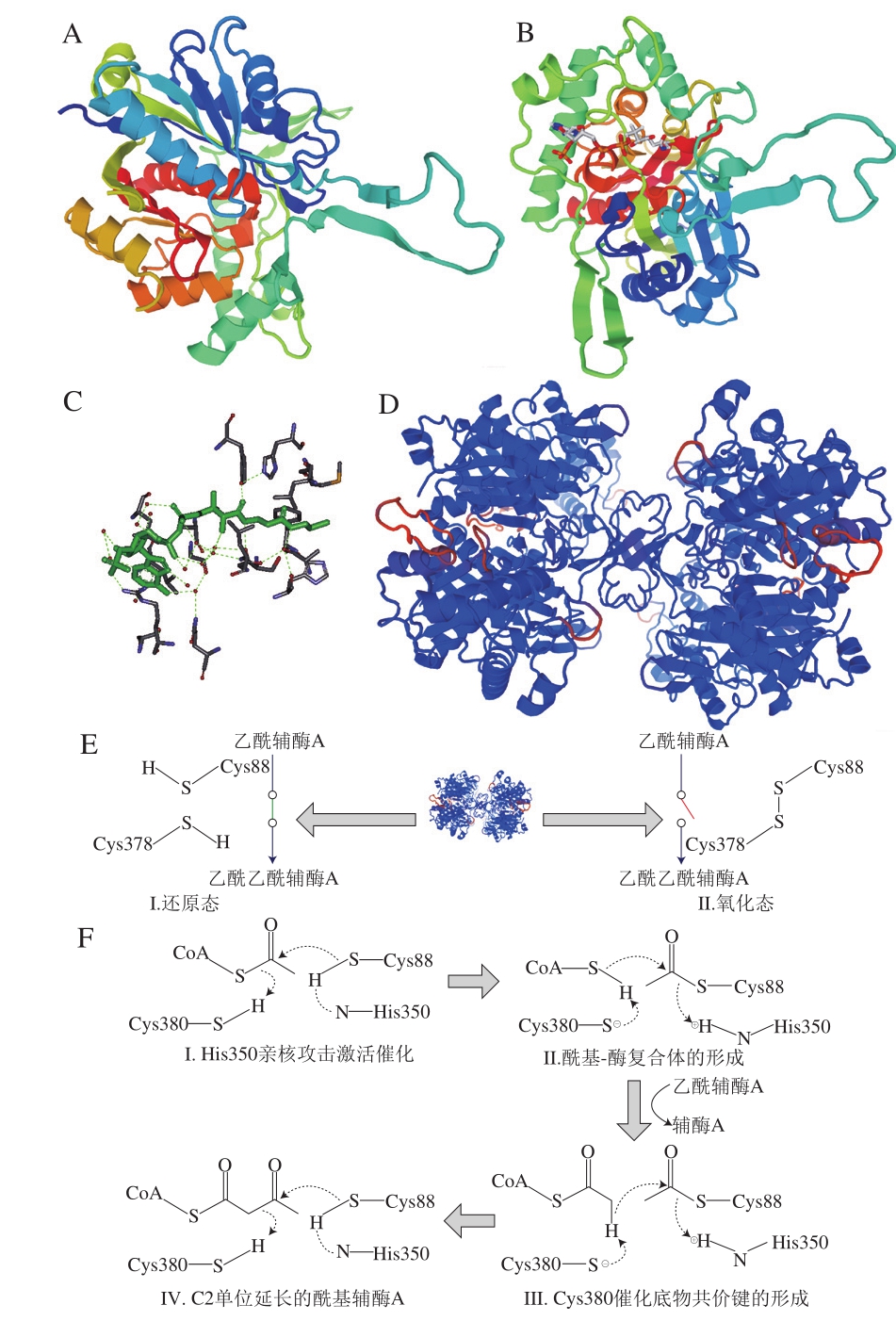
图7 关键酶ThlA1单体的三维结构模拟(A)、分子对接(B)、酶-底物作用关系分析(C)、四聚体结构预测(D)、氧化还原开关催化调控(E)和具体催化机制(F)
Fig. 7 Monomer modeling (A), molecular docking (B), enzymesubstrate interaction analysis (C), tetramer structure prediction (D),redox-switch catalytic regulation (E) and catalytic mechanism (F) of the key enzyme ThlA1
基于结构解析和分子对接,推测ThlA1的催化机制如下:His350通过亲核攻击并转移Cys88与硫原子连接的氢离子激活Cys88,Cys88的硫原子与酰基辅酶A的羰基碳原子作用生成共价键,形成酰基-酶复合体,同时Cys380与硫原子连接的氢离子转移到失去酰基的辅酶A上(图7F I和II)。之后,失去氢离子的Cys380与另一分子的酰基辅酶A发生作用,获取底物酰基的氢离子,并激活底物的甲基碳原子向酰基-酶复合体的羰基碳原子进行亲核攻击,形成共价键,获得以C2单位延长的酰基辅酶A(图7F III和IV)[23-24,28,30]。碳链延长的酰基辅酶A最终经硫酯酶的催化作用脱去辅酶A生成对应链长的脂肪酸[11](图1)。
由于己酸对中国传统酿造浓香型白酒风味与品质的重要作用和C. kluyveri对己酸合成的突出贡献,研究对已知遗传信息的3 株C. kluyveri进行比较基因组学分析和己酸代谢途径关键基因的深度挖掘及重新注释。通过代谢途径分析结合文献研究,聚焦于C. kluyveri DSM 555来源己酸代谢途径关键基因编码的硫解酶ThlA1,对其序列和结构进行全面的生物信息学分析,发现C. kluyveri来源ThlA1具有独特的氧化还原开关调控机制,并结合分子对接,对具体催化的过程进行阐释。上述研究对于白酒重要功能微生物C. kluyveri己酸代谢合成途径的科学解析,以及基于此改造菌株的己酸代谢性能,进而科学调控传统浓香型白酒的酿造具有重要意义。
[1] XU Y, SUN B, FAN G, et al. The brewing process and microbial diversity of strong flavour Chinese spirits: a review[J]. Journal of the Institute of Brewing, 2017, 123: 5-12. DOI:10.1002/jib.404.
[2] 孙宝国, 吴继红, 黄明泉, 等. 白酒风味化学研究进展[J]. 中国食品学报, 2015, 15(9): 1-8. DOI:10.16429/j.1009-7848.2015.09.001.
[3] 康永璞, 郭新光, 刘凤翔, 等. 浓香型白酒: GB/T 10781.1—2006[S].北京: 中国标准出版社, 2006.
[4] 王晓丹, 胥思霞, 班世栋, 等. 红曲酯化酶粗酶制剂在浓香型青酒大曲酒生产中的应用研究[J]. 酿酒科技, 2014(7): 57-60.DOI:10.13746/j.njkj.2014.0093.
[5] 李绍亮, 李亚凤, 李学思. 酿酒大曲中酯化红曲的分离及应用[J].酿酒, 2014, 41(2): 32-39. DOI:10.3969/j.issn.1002-8110.2014.02.010.[6] 蒋育萌, 吴成全. 酯化红曲在浓香型白酒生产中的应用[J]. 酿酒科技, 2011(12): 70-72. DOI:10.13746/j.njkj.2011.12.031.
[7] ZHU X, TAO Y, LIANG C, et al. The synthesis of n-caproate from lactate: a new efficient process for medium-chain carboxylates production[J]. Scientific Reports, 2015, 5: 14360. DOI:10.1038/srep14360.
[8] SEEDORF H, FRICKE W F, VEITH B, et al. The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features[J]. Proceedings of the National Academy of Sciences, 2008,105(6): 2128-2133. DOI:10.1073/pnas.0711093105.
[9] VASUDEVAN D, RICHTER H, ANGENENT L T. Upgrading dilute ethanol from syngas fermentation to n-caproate with reactor microbiomes[J]. Bioresource Technology, 2014, 151: 378-382.DOI:10.1016/j.biortech.2013.09.105.
[10] TAO Y, ZHU X, WANG H, et al. Complete genome sequence of Ruminococcaceae bacterium CPB6: a newly isolated culture for efficient n-caproic acid production from lactate[J]. Journal of Biotechnology, 2017, 259: 91-94. DOI:10.1016/j.jbiotec.2017.07.036.
[11] CHEON Y, KIM J S, PARK J B, et al. A biosynthetic pathway for hexanoic acid production in Kluyveromyces marxianus[J]. Journal of Biotechnology, 2014, 182: 30-36. DOI:10.1016/j.jbiotec.2014.04.010.
[12] KIM S G, JANG S, LIM J H, et al. Optimization of hexanoic acid production in recombinant Escherichia coli by precise flux rebalancing[J]. Bioresource Technology, 2018, 247: 1253-1257.DOI:10.1016/j.biortech.2017.10.014.
[13] XIAO Y, FRANCKE C, ABEE T, et al. Clostridial spore germination versus bacilli: genome mining and current insights[J]. Food Microbiology, 2011, 28(2): 266-274. DOI:10.1016/j.fm.2010.03.016.
[14] DARLING A C E, MAU B, BLATTNER F R, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements[J].Genome Research, 2004, 14(7): 1394-1403. DOI:10.1101/gr.2289704.
[15] GASTEIGER E, HOOGLAND C, GATTIKER A, et al. Protein identification and analysis tools on the ExPASy server[M]//WALKER J M. The proteomics protocols handbook. Clifton: Humana Press,2005: 571-607. DOI:10.1385/1-59259-890-0:571.
[16] TAMURA K, PETERSON D, PETERSON N, et al. MEGA5:molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance, and maximum parsimony methods[J]. Molecular Biology and Evolution, 2011, 28(10): 2731-2739. DOI:10.1093/molbev/msr121.
[17] NIELSEN H. Predicting secretory proteins with signal P[J]. Methods in Molecular Biology, 2017, 1611: 59-73. DOI:10.1007/978-1-4939-7015-5_6.
[18] GEOURJON C, DELEAGE G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments[J]. Bioinformatics, 1995, 11(6): 681-684.DOI:10.1093/bioinformatics/11.6.681.
[19] ARNOLD K, BORDOLI L, KOPP J, et al. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling[J]. Bioinformatics, 2006, 22(2): 195-201. DOI:10.1093/bioinformatics/bti770.
[20] GAO Y D, HUANG J F. An extension strategy of Discovery Studio 2.0 for non-bonded interaction energy automatic calculation at the residue level[J]. Zoological Research, 2011, 32(3): 262-266. DOI:10.3724/SP.J.1141.2011.03262.
[21] JAIN R, RIVERA M C, LAKE J A. Horizontal gene transfer among genomes: the complexity hypothesis[J]. Proceedings of the National Academy of Sciences, 1999, 96(7): 3801-3806. DOI:10.1073/pnas.96.7.3801.
[22] GURUPRASAD K, REDDY B V, PANDIT M W. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence[J]. Protein Engineering, Design and Selection, 1990, 4(2):155-161. DOI:10.1093/protein/4.2.155.
[23] HAAPALAINEN A M, MERILÄINEN G, WIERENGA R K. The thiolase superfamily: condensing enzymes with diverse reaction specificities[J]. Trends in Biochemical Sciences, 2006, 31(1): 64-71.DOI:10.1016/j.tibs.2005.11.011.
[24] MODIS Y, WIERENGA R K. Crystallographic analysis of the reaction pathway of Zoogloea ramigera biosynthetic thiolase[J]. Journal of Molecular Biology, 2000, 297(5): 1171-1182. DOI:10.1006/jmbi.2000.3638.
[25] GASTEIGER E, HOOGLAND C, GATTIKER A, et al. The proteomics protocols handbook: protein identification and analysis tools on the ExPASy server[M]. Clifton: Humana Press, 2005: 571-607. DOI:10.1385/1-59259-890-0:571.
[26] 白姝, 常颖, 刘小娟, 等. 海藻糖和氨基酸之间相互作用的分子动力学模拟[J]. 物理化学学报, 2014, 30(7): 1239-1246. DOI:10.3866/PKU.WHXB201405151.
[27] HARTL F U, BRACHER A, HAYER-HARTL M. Molecular chaperones in protein folding and proteostasis[J]. Nature, 2011, 475:324-332. DOI:10.1038/nature10317.
[28] KIM S, JANG Y S, HA S C, et al. Redox-switch regulatory mechanism of thiolase from Clostridium acetobutylicum[J]. Nature Communications, 2015, 6: 8410. DOI:10.1038/ncomms9410.
[29] DROZDETSKIY A, COLE C, PROCTER J, et al. JPred4: a protein secondary structure predicition server[J]. Nucleic Acids Research,2015, 43(W1): 389-394. DOI:10.1093/nar/gkv332.
[30] HEATH R J, ROCK C O. The claisen condensation in biology[J].Natural Product Reports, 2002, 19(5): 581-596. DOI:10.1039/b110221b.
Bioinformatics Analysis of the Key Enzymes of the Hexanoic Acid Metabolic Pathway in Clostridium kluyveri Based on Comparative Genomics
XU Youqiang, SUN Baoguo, JIANG Yuefeng, et al. Bioinformatics analysis of the key enzymes of the hexanoic acid metabolic pathway in Clostridium kluyveri based on comparative genomics[J]. Food Science, 2019, 40(4): 122-129.(in Chinese with English abstract) DOI:10.7506/spkx1002-6630-20180413-183. http://www.spkx.net.cn